一种卤化效应功能化的手性水凝胶材料及其制备方法和应用
1.本发明属于超分子化学领域,涉及一种卤化效应功能化的手性水凝胶材料及其制备方法和应用。
背景技术:
2.手性是自然界最重要的化学信号之一,在遗传信息的复制、转录及蛋白质的高级结构的形成、蛋白质功能化方面有着重要影响。对保持人体组织细胞的正常功能起到了决定作用。π-π堆积、氢键、静电相互作用和范德华力等分子间力是自然体系将生物体的基本单元的分子手性转移和放大为超分子手性结构的关键决定因素。构建手性微纳米结构,探索手性影响细胞行为的内在机理不仅在生物医药领域有应用价值,也对探讨自然界生命的手性起源具有重大意义。近年来,国内外学者对分子手性结构的构建及超分子手性组装策略进行了研究,开发出了许多生物相容性好且功能多样化的手性组装体。尽管如此,关于超分子手性组装体的构建仍旧处于前期的探索阶段,还存在很多亟需解决的关键科学问题。其中,现有的手性组装体形成是大多通过分子修饰,组装环境(ph、溶剂和温度)的变化以及共组装来介导非共价相互作用实现分子手性信号的放大和调控超分子手性反转,以至于现有的手性结构在复杂性和功能性上与原生的手性结构相差很远,且很难获得手性均一的手性组装体。因此,需要新的作用力和组装策略来推动更复杂的超分子材料。
3.卤化效应,指将卤素原子(f、cl、br或i)引入有机化合物中,因卤素原子独特的化学性质使化合物或者材料的某一特性发生变化。其中有机卤化物中的共价卤素原子可以作为路易斯酸与中性的或者带负电的路易斯碱之间形成的非共价相互作用,这是一种类似氢键的分子间弱相互作用。而氟原子具有电负性强,原子半径小等独特的内在性质,使氟原子不仅可以参与卤键的形成,还可以作为氢键的受体参与氢键的形成等多种共价相互作用的形成。卤素原子参与的非共价相互作用在晶体工程、功能材料设计、药物设计等很多领域有着广泛的应用,但卤化效应在控制超分子手性结构影响的报道很少。
技术实现要素:
4.针对上述技术问题,本发明提出一种卤化效应功能化的手性水凝胶材料及其制备方法和应用。本发明利用不同卤素原子取代的卤化效应,或相同卤素原子不同取代位置的卤化效应改变化学分子的空间构象,使其卤化效应功能化的手性水凝胶材料的手性出现相对应的改变,得到了由单一对映体构建的不同手性的螺旋纤维结构。将其用于细胞粘附增殖时,具有突出的效果。且本发明提供的制备方法无需复杂的反应,易于规模化生产,所得产物纯度高;手性纳米纤维水凝胶调控细胞的黏附行为均在生理环境下进行,操作方便,具有实际应用价值。
5.为了达到上述目的,本发明的技术方案是这样实现的:
6.一种卤化效应功能化的手性水凝胶材料,通过引入不同卤素原子及变换卤素原子取代位置或侧链-亲疏水性改变凝胶因子空间构象,实现由单一手性分子构建不同螺旋手
性、具有卤化效应功能化的手性水凝胶材料,同时凝胶因子通过氢键、芳香堆积作用、氟氢键和卤键物理相互作用进行自组装。
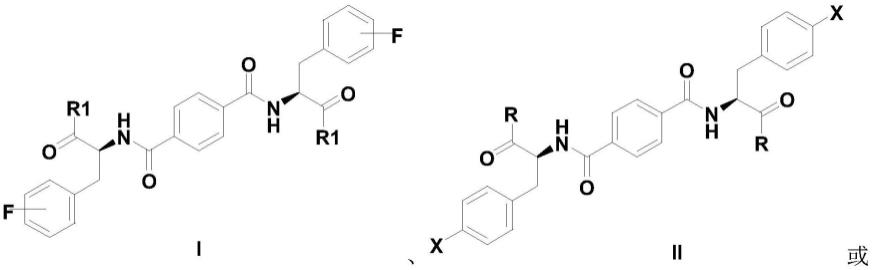
7.进一步,所述凝胶因子为l-苯丙氨酸衍生物,所述卤化效应功能化的手性水凝胶材料的结构式如i、ii或iii所示:料的结构式如i、ii或iii所示:其中,-f表示f取代位置为邻位、间位或对位中的任意一种;r1为r为x为氟、氯、溴或碘中的任意一种。
8.进一步,上述卤化效应功能化的手性水凝胶材料的制备方法,为:
9.(1)当卤化效应功能化的手性水凝胶材料为i所示的结构式时,其制备步骤如下:
10.a1、将氟取代的boc-l-苯丙氨酸和1-羟基苯并三唑(hobt)用二氯甲烷溶解,冰浴冷却,加入二甘醇胺和n,n-二异丙基乙胺(dipea),冰浴30min后,加入edci;将反应溶液缓慢升至室温,继续室温反应12h;依次用饱和柠檬酸溶液,饱和碳酸氢钠溶液,饱和氯化钠溶液洗涤,旋干,用乙酸乙酯和石油醚重结晶,得到boc-l-nf-phe,其结构式为-f表示f取代位置为邻位、间位或对位中的任意一种;
11.a2、将步骤a1所得的boc-l-nf-phe加入到二氯甲烷与三氟乙酸的混合溶液中,室温反应3h后,脱boc-保护基,将溶液旋转蒸发完全除去溶液和剩余l-nf-phe的三氟乙酸,得
到l-nf-phe三氟乙酸盐,其结构式为-f表示f取代位置为邻位、间位或对位中的任意一种;
12.a3、将步骤a2所得的l-nf-phe三氟乙酸盐溶于二氯甲烷,冰浴冷却,依次加入三乙胺、对苯二甲酰氯,将反应溶液缓慢升至室温并搅拌,室温反应24h,反应结束后将所得胶体抽滤,二氯甲烷、乙醇、去离子水依次洗涤,干燥样品,得到白色固体目标产物i,其结构式为-f表示f取代位置为邻位、间位或对位中的任意一种。
13.进一步,当卤化效应功能化的手性水凝胶材料为i所示的结构式时,所述步骤a1中氟原子取代的boc-l-苯丙氨酸和二甘醇胺在n,n-二异丙基乙胺(dipea),1-羟基苯并三唑(hobt)和edci存在下发生酰胺缩合反应。
14.进一步,所述步骤a1中氟取代的boc-l-苯丙氨酸结构式为-f表示f取代位置为邻位、间位或对位中的任意一种,所述二甘醇胺的结构上为
15.进一步,所述步骤a1中氟取代的boc-l-苯丙氨酸与二甘醇胺、n,n-二异丙基乙胺、1-羟基苯并三唑、edci的摩尔比为1:1.5:3.6:1.3:2。
16.进一步,所述步骤a2中二氯甲烷与三氟乙酸的混合溶液中二氯甲烷与三氟乙酸的体积比为(1-4):1。
17.进一步,所述步骤a3中对苯二甲酰氯的结构式为对苯二甲酰氯与l-nf-phe三氟乙酸盐和三乙胺的摩尔比为1:(2.1~2.3):(10~15)。
18.进一步,所述步骤a3中二氯甲烷、乙醇、去离子水依次洗涤的用量分别为30~
50ml、10~20ml、30~50ml。
19.(2)当卤化效应功能化的手性水凝胶材料为ii所示的结构式时,其制备步骤如下:
20.b1、在氮气保护,冰浴条件下,将二氯亚砜缓慢滴加到干燥的甲醇溶液中,10min后,反应液缓慢升至室温,加入卤素原子-l-苯丙氨酸,室温过夜反应,旋干,得到甲酯保护的卤代l-苯丙氨酸,其结构式为x为氟、氯、溴或碘中的任意一种;
21.b2、将步骤b1所得的甲酯保护的卤代l-苯丙氨酸溶于二氯甲烷,冰浴冷却,依次加入三乙胺、对苯二甲酰氯,将反应溶液缓慢升至室温并搅拌,室温反应24h,反应结束后将所得胶体抽滤,二氯甲烷、乙醇、去离子水依次洗涤,干燥样品,得到白色固体l-4x-phe-ome,其结构式为x为氟、氯、溴或碘中的任意一种;
22.b3、将步骤b2所得的l-4x-phe-ome溶于甲醇中,再加入2m的氢氧化钠溶液,室温反应24h后,用1m的盐酸溶液调节ph至2~3,将所得胶体抽滤,去离子水洗涤,干燥样品,得到白色固体l-4x-phe-oh,其结构式为x为氟、氯、溴或碘中的任意一种;
23.b4-1、将步骤b3所得的l-4x-phe-oh和1-羟基苯并三唑用二氯甲烷溶解,冰浴冷却,加入二甘醇胺和n,n-二异丙基乙胺,冰浴30min后,加入edci;将反应溶液缓慢升至室温并搅拌,室温反应12h,反应结束后将所得胶体抽滤,二氯甲烷、乙醇、去离子水依次洗涤,干燥样品,得到白色固体目标产物ii l-4x-phe,其结构式为
x为氟、氯、溴或碘中的任意一种。
24.b4-2、向步骤b3所得的l-4x-phe-oh与5倍当量二甘醇的混合物中滴加0.5ml滴浓盐酸,升温至135℃,反应4h,反应完成后,缓慢滴入去离子水中将所得胶体抽滤,去离子水洗涤,干燥样品,得到白色固体目标产物ii l-4x-es,其结构式为x为氟、氯、溴或碘中的任意一种。
25.进一步,当卤化效应功能化的手性水凝胶材料为ii所示的结构式时,所述步骤b1中的卤素原子-l-苯丙氨酸的结构式为x为氟、氯、溴或碘中的任意一种。
26.进一步,所述步骤b1中卤素原子-l-苯丙氨酸与二氯亚砜的摩尔比为1:3。
27.进一步,所述步骤b2中对苯二甲酰氯的结构式为对苯二甲酰氯与甲酯保护的卤代l-苯丙氨和三乙胺的摩尔比为1:(2.1~2.3):(10~15)。
28.进一步,所述步骤b3中甲醇与2m氢氧化钠溶液的体积比为3:1。
29.进一步,所述步骤b4-1中二甘醇胺的结构上为l-4x-phe-oh与二甘醇胺、n,n-二异丙基乙胺、1-羟基苯并三唑、edci的摩尔比为1:3:7.2:2.6:4,二氯甲烷、乙醇、去离子水依次洗涤的用量分别为30~50ml、10~20ml、30~50ml。
30.(3)当卤化效应功能化的手性水凝胶材料为iii所示的结构式时,其制备步骤如下:
31.c1、在氮气保护,冰浴条件下,将二氯亚砜缓慢滴加到干燥的甲醇溶液中,10min
后,反应液缓慢升至室温,加入卤素原子-l-苯丙氨酸,室温反应12h。旋干,得到甲酯保护的卤代l-苯丙氨酸x为氟、氯、溴或碘中的任意一种;
32.c2、将对苯二乙酸和1-羟基苯并三唑用二氯甲烷溶解,冰浴冷却,加入n,n-二异丙基乙胺和步骤c1所得的甲酯保护的卤代l-苯丙氨酸,冰浴30min后,加入edci;将反应溶液缓慢升至室温并搅拌,室温反应12h,将所得胶体布氏漏斗抽滤,二氯甲烷、乙醇、去离子水依次洗涤,烘箱干燥样品,得到白色固体l-4x-pa-ome,其结构式为x为氟、氯、溴或碘中的任意一种;
33.c3、将步骤c2所得的l-4x-pa-ome溶于甲醇中,再加入2m的氢氧化钠溶液,室温反应24h后,用1m的盐酸溶液调节ph为2~3,所得胶体布氏漏斗抽滤,去离子水洗涤,烘箱干燥样品,得到白色固体l-4x-pa-oh,其结构式为x为氟、氯、溴或碘中的任意一种;
34.c4、将步骤c3所得的l-4x-pa-oh与1-羟基苯并三唑用二氯甲烷溶解,冰浴冷却,加入二甘醇胺和n,n-二异丙基乙胺,冰浴30min后,加入edci;将反应溶液缓慢升至室温并搅拌,室温反应12h,将所得胶体布氏漏斗抽滤,二氯甲烷、乙醇、去离子水依次洗涤,烘箱干燥样品,得到白色固体目标产物iii l-4x-pa,其结构式为
35.x为氟、氯、溴或碘中的任意一种。
36.进一步,当卤化效应功能化的手性水凝胶材料为iii所示的结构式时,所述步骤c1
中的卤素原子-l-苯丙氨酸的结构式为x为氟、氯、溴或碘中的任意一种。
37.进一步,所述步骤c1中卤素原子-l-苯丙氨酸与二氯亚砜的摩尔比为1:3。
38.进一步,步骤c1中卤素原子取代的l-苯丙氨酸在二氯亚砜和甲醇存在下发生酯化反应,生成甲酯保护的卤代l-苯丙氨酸。
39.进一步,所述步骤c2中甲酯保护的卤代l-苯丙氨酸与对苯二乙酸、n,n-二异丙基乙胺、1-羟基苯并三唑、edci的摩尔比为2:1:7.2:2.6:4,二氯甲烷、乙醇、去离子水依次洗涤的用量分别为30~50ml、10~20ml、30~50ml。
40.进一步,步骤c2中甲酯保护的卤代l-苯丙氨酸和对苯二乙酸在n,n-二异丙基乙胺,1-羟基苯并三唑和edci存在下发生酰胺缩合反应,生成l-4x-pa-ome。
41.进一步,所述步骤c3中甲醇与2m氢氧化钠溶液的体积比为3:1。
42.进一步,在步骤c3中,在甲醇、2m氢氧化钠溶液存在的条件下,l-4x-pa-ome脱去甲酯,生成了l-4x-pa-oh。
43.进一步,所述步骤c4中二甘醇胺的结构上为l-4x-pa-oh与二甘醇胺、n,n-二异丙基乙胺、1-羟基苯并三唑、edci的摩尔比为1:3:7.2:2.6:4。
44.进一步,所述的卤化效应功能化的手性水凝胶材料通过分子手性和组装纤维手性在促进细胞黏附、增殖中的应用。
45.进一步,所述的细胞为hep g2细胞。
46.进一步,所述的细胞为nih 3t3细胞。
47.本发明具有以下有益效果:
48.1、本发明是利用不同卤素原子及其取代位置或侧链亲疏水来调控纳米纤维的螺旋手性,与传统利用对映体分子、溶剂、温度、客体分子、光照和ph等变量引起螺旋结构手性的反转相比,明显改进了现有体系的生物相容性差、结构不稳定以及操作困难等缺点。
49.2、本发明由单一对映体构建的手性纳米纤维材料通过分子手性和组装纤维手性的变化调控细胞黏附行为,揭示了手性纳米纤维调控细胞行为过程中分子手性和超分子手性的作用。
50.3、本发明所制备的卤化效应功能化的手性水凝胶材料具有合成简单、良好生物相容性、可降解的优点,并且是由单一对映体构建的手性纳米纤维,手性易于调控,可望在细胞组织培养和其他医学领域有独特的应用。
51.4、本发明中卤化效应功能化的手性水凝胶材料的制备方法无需复杂的反应,易于规模化生产,所得产物纯度高;手性纳米纤维水凝胶调控细胞的黏附行为均在生理环境下进行,操作方便,具有实际应用价值。
52.5、将本发明制备的卤化效应功能化的手性水凝胶材料应用于细胞黏附、增殖,特
别是hep g2细胞和nih 3t3细胞的黏附、增殖时,nih 3t3细胞黏附在l-4f-phe(lm)纳米纤维膜上的数量是d-4f-phe(dp)膜上的2.2倍,是l-phe(lp)膜上的1.6倍,是d-phe(dm)膜上的1.3倍,是ps孔上的1.75倍。hepg2细胞黏附在l-4f-phe(lm)膜上的细胞数量分别是d-4f-phe(dp)膜上、l-phe(lp)膜上、d-phe(dm)膜上和ps孔上的细胞数量的1.9倍、1.3倍、1.1倍和1.5倍。
附图说明
53.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
54.图1为本发明实施例1制备的卤化效应功能化的手性水凝胶材料的图片,其中,a为氟原子不同位置取代功能化的水凝胶图片,b和c为不同卤素原子功能化的水凝胶图片;
55.图2为本发明实施例1制备的l-phe、l-2f-phe、l-3f-phe、l-4f-phe的超分子水凝胶的纳米纤维扫描比较图;其中,l-phe和l-2f-phe为右手螺旋,l-3f-phe和l-4f-phe为左手螺旋。
56.图3为本发明nih 3t3细胞和hepg2细胞在本发明卤化效应功能化的手性纳米纤维l-phe、l-4f-phe、d-phe和d-4f-phe薄膜上孵育不同时间后的荧光显微镜照片及细胞黏附密度;其中,a)为l-phe、l-4f-phe、d-phe和d-4f-phe纳米纤维薄膜以及ps对照板上nih 3t3细胞培养3天后荧光照片以及孵育1天,2天,3天后,l-phe、l-4f-phe、d-phe和d-4f-phe纳米纤维膜和ps对照的nih 3t3细胞的定量数据,n=6;*,**,***数据显示显着差异(anova:*p0.05,**p0.005,***p0.001);b)为l-phe、l-4f-phe、d-phe和d-4f-phe纳米纤维薄膜以及ps对照板上hepg2细胞培养3天后荧光照片以及孵育1天,2天,3天后,l-phe、l-4f-phe、d-phe和d-4f-phe纳米纤维膜和ps对照的hepg2细胞的定量数据,n=6;*,**,***数据显示显著差异(anova:*p0.05,**p0.005,***p0.001);c)为手性水凝胶三维细胞培养3天后的共聚焦显微镜照片。
57.图4为本发明实施例1制备的氟化功能化的手性水凝胶材料的圆二色图谱。
58.图5为本发明实施例1制备的手性水凝胶材料l-phe的1hnmr谱。
59.图6为本发明实施例1制备的氟化效应功能化的手性水凝胶材料l-2f-phe的1h nmr谱。
60.图7为本发明实施例1制备的氟化效应功能化的手性水凝胶材料l-3f-phe的1h nmr谱。
61.图8为本发明实施例1制备的氟化效应功能化的手性水凝胶材料l-4f-phe的1h nmr谱。
62.图9为本发明实施例1制备的卤代效应功能化的手性水凝胶材料l-4cl-phe的1h nmr谱。
63.图10为本发明实施例1制备的卤代效应功能化的手性水凝胶材料l-4br-phe的1h nmr谱。
64.图11为本发明实施例1制备的卤代效应功能化的手性水凝胶材料l-4i-phe的
4f-phe)。
78.b部分中,均为l-苯丙氨酸衍生物,其中r为nh2(ch2)2o(ch2)2oh(l-4x-phe)或ho(ch2)2o(ch2)2oh(l-4x-es);x为f、cl、br、i。
79.c部分中,均为l-苯丙氨酸衍生物,其中r1为nh2(ch2)2o(ch2)2oh(l-4x-pa);x为f、cl、br、i。
80.a部分中,是利用氟原子的取代位置不同,实现了超分子组装体的手性反转可控。b部分中,是利用不同卤素原子的取代不同,实现了超分子组装体的手性反转可控。c部分中,是利用不同卤素原子的取代不同,实现了超分子组装体的手性反转可控。手性凝胶因子在自组装过程中,分子手性信号将传递到纳米纤维组装体结构中,得到类似人体内胶原蛋白螺旋结构的手性凝胶纤维。a部分的l-苯丙氨酸衍生物,当氟原子在邻位时(l-2f-phe),可得到右手螺旋纳米纤维;当氟原子在间位(l-3f-phe)和对位(l-4f-phe)时,可得到左手螺旋纳米纤维;d-苯丙氨酸衍生物部分,氟原子在对位(d-4f-phe)时,可得到右手螺旋纳米纤维。
81.所述卤代效应功能化的手性水凝胶材料的制备方法,步骤如下:
82.(1)a部分凝胶因子合成步骤:
83.步骤a1,boc-l-nf-phe的制备
[0084][0085]
boc-l-nf-phe的制备:将氟取代的boc-l-苯丙氨酸和1-羟基苯并三唑用二氯甲烷溶解,冰浴冷却,加入二甘醇胺和n,n-二异丙基乙胺,冰浴30min后,加入edci;将反应溶液缓慢升至室温,继续室温反应12h;依次用饱和柠檬酸溶液,饱和碳酸氢钠溶液,饱和氯化钠溶液洗涤,旋干,用乙酸乙酯和石油醚重结晶,其中氟取代的boc-l-苯丙氨酸与二甘醇胺、n,n-二异丙基乙胺、1-羟基苯并三唑、edci的摩尔比为1:1.5:3.6:1.3:2。
[0086]
步骤a2,l-nf-phe三氟乙酸盐的合成
[0087][0088]
boc-l-nf-phe加入到二氯甲烷/三氟乙酸体积比为4/1的溶液中,室温反应3h后,将溶液旋转蒸发完全除去溶液和剩余l-nf-phe的三氟乙酸。
[0089]
步骤a3,氟原子不同取代位置凝胶因子的制备:
[0090][0091]
将l-nf-phe的三氟乙酸盐溶于二氯甲烷,冰浴冷却,依次加入三乙胺、对苯二甲酰氯,将反应溶液缓慢升至室温并搅拌,室温反应24h。将所得胶体布氏漏斗抽滤,二氯甲烷、
乙醇、去离子水依次洗涤,烘箱干燥样品,得到白色固体目标产物i,对苯二甲酰氯与l-nf-phe三氟乙酸盐和三乙胺的摩尔比为1:2.2:10
[0092]
根据氟的取代位置的不同,可选用邻位、间位、对位氟取代的boc-l-苯丙氨酸,由此根据上述条件分别制得l-2f-phe、l-3f-phe和l-4f-phe。
[0093]
选择氟的对位取代的boc-d-苯丙氨酸,由此根据上述条件制得d-4f-phe。
[0094]
(2)b部分凝胶因子合成步骤:
[0095]
步骤b1,甲酯保护的卤代l-苯丙氨酸的合成
[0096][0097]
在氮气保护,冰浴条件下,将二氯亚砜缓慢滴加到干燥的甲醇溶液中,10min后,反应液缓慢升至室温,加入卤素原子-l-苯丙氨酸,室温反应12h。旋干即可进行下一步反应,卤素原子-l-苯丙氨酸与二氯亚砜的摩尔比为1:3。
[0098]
步骤b2,l-4x-phe-ome的合成
[0099][0100]
将甲酯保护的卤代l-苯丙氨酸溶于二氯甲烷,冰浴冷却,依次加入三乙胺、对苯二甲酰氯,对苯二甲酰氯,甲酯保护的卤代l-苯丙氨酸、三乙胺的摩尔比为1:2.2:10将反应溶液缓慢升至室温并搅拌,室温反应24h。将所得胶体布氏漏斗抽滤,100ml二氯甲烷、100ml乙醇、500ml去离子水依次洗涤,烘箱干燥样品,得到白色固体l-4x-phe-ome。
[0101]
步骤b3,l-4x-phe-oh的制备:
[0102][0103]
将l-4x-phe-ome溶于甲醇中,再加入2m的氢氧化钠溶液,甲醇与2m氢氧化钠溶液的体积比为3:1室温反应24h后,用1m的盐酸溶液调节ph为2~3,所得胶体布氏漏斗抽滤,去离子水洗涤,烘箱干燥样品,得到白色固体l-4x-phe-oh。
[0104]
步骤b4,不同卤素原子功能化凝胶因子的制备:
[0105][0106]
b4-1)将l-4x-phe-oh和1-羟基苯并三唑用二氯甲烷溶解,冰浴冷却,加入二甘醇胺和n,n-二异丙基乙胺,冰浴30min后,加入edci;将反应溶液缓慢升至室温并搅拌,室温反应12h,将所得胶体布氏漏斗抽滤,二氯甲烷、乙醇、去离子水依次洗涤,烘箱干燥样品,得到白色固体目标产物ii l-4x-phe,l-4x-phe-oh与二甘醇胺、n,n-二异丙基乙胺、1-羟基苯并三唑、edci的摩尔比为1:3:7.2:2.6:4。
[0107]
当时,根据卤素原子-l-苯丙氨酸对位上卤原子的不同,当卤原子分别为cl、br、i时,可选用cl取代、br取代、i取代的l-苯丙氨酸,由此根据上述条件分别制得l-4cl-phe、l-4br-phe、l-4i-phe。
[0108]
b4-2)向l-4x-phe-oh与二甘醇的混合物中滴加浓盐酸,升温至135℃,反应4h,反应完成后,缓慢滴入去离子水中将所得胶体抽滤,去离子水洗涤,烘箱干燥样品,得到白色固体目标产物iil-4x-es。
[0109]
当时,根据卤素原子-l-苯丙氨酸对位上卤原子的不同,当卤原子分别为f、cl、br、i时,可选用f取代、cl取代、br取代、i取代的l-苯丙氨酸,由此根据上述条件分别制得l-4f-es、l-4cl-es、l-4br-es、和l-4i-es。
[0110]
(3)c部分凝胶因子合成步骤:
[0111]
步骤c1,甲酯保护的卤代l-苯丙氨酸的合成
[0112][0113]
在氮气保护,冰浴条件下,将二氯亚砜缓慢滴加到干燥的甲醇溶液中,10min后,反应液缓慢升至室温,加入卤素原子-l-苯丙氨酸,卤素原子-l-苯丙氨酸与二氯亚砜的摩尔比为1:3室温反应12小时。旋干即可进行下一步反应。
[0114]
步骤c2,l-4x-pa-ome的合成
[0115][0116]
将对苯二乙酸和1-羟基苯并三唑用二氯甲烷溶解,冰浴冷却,加入,n-二异丙基乙胺和甲酯保护的卤代l-苯丙氨酸,冰浴30min后,加入edci;将反应溶液缓慢升至室温并搅拌,室温反应12h,将所得胶体布氏漏斗抽滤,二氯甲烷、乙醇、去离子水依次洗涤,烘箱干燥样品,得到白色固体l-4x-pa-ome,甲酯保护的卤代l-苯丙氨酸与对苯二乙酸与n,n-二异丙基乙胺,1-羟基苯并三唑,edci的摩尔比为2:1:7.2:2.6:4。
[0117]
步骤c3,l-4x-pa-oh的制备:
[0118][0119]
将l-4x-pa-ome溶于甲醇中,再加入2m的氢氧化钠溶液,甲醇与2m氢氧化钠溶液的体积比为3:1室温反应24h后,用1m的盐酸溶液调节ph为2~3,所得胶体布氏漏斗抽滤,去离子水洗涤,烘箱干燥样品,得到白色固体l-4x-pa-oh。
[0120]
步骤c4,不同卤素原子功能化凝胶因子的制备:
[0121][0122]
所述l-4x-pa-oh和1-羟基苯并三唑用二氯甲烷溶解,冰浴冷却,加入二甘醇胺和n,n-二异丙基乙胺,冰浴30min后,加入edci;l-4x-pa-oh与二甘醇胺、n,n-二异丙基乙胺、1-羟基苯并三唑、edci的摩尔比为1:3:7.2:2.6:4,将反应溶液缓慢升至室温并搅拌,室温反应12h,将所得胶体布氏漏斗抽滤,二氯甲烷、乙醇、去离子水依次洗涤,烘箱干燥样品,得到白色固体目标产物l-4x-pa。
[0123]
根据卤素原子-l-苯丙氨酸对位上卤原子的不同,当卤原子分别为f、cl、br、i时,可选用f取代、cl取代、br取代、i取代的l-苯丙氨酸,由此根据上述条件分别制得l-4f-pa、l-4cl-pa、l-4br-pa、和l-4i-pa。
[0124]
对比例1
[0125]
本对比例为l-phe的制备方法,步骤如下:
[0126]
步骤a1,boc-l-n-phe的制备
[0127][0128]
boc-l-n-phe的制备:将boc-l-苯丙氨酸和1-羟基苯并三唑用二氯甲烷溶解,冰浴冷却,加入二甘醇胺和n,n-二异丙基乙胺,冰浴30min后,加入edci;将反应溶液缓慢升至室温,继续室温反应12h;依次用饱和柠檬酸溶液,饱和碳酸氢钠溶液,饱和氯化钠溶液洗涤,
旋干,用乙酸乙酯和石油醚重结晶,其中boc-l-苯丙氨酸与二甘醇胺、n,n-二异丙基乙胺、1-羟基苯并三唑、edci的摩尔比为1:1.5:3.6:1.3:2。
[0129]
步骤a2,l-n-phe三氟乙酸盐的合成
[0130][0131]
boc-l-nf-phe加入到二氯甲烷/三氟乙酸(v/v)为4/1的溶液中,室温反应3h后,将溶液旋转蒸发完全除去溶液和剩余l-n-phe的三氟乙酸。
[0132]
步骤a3,氟原子不同取代位置凝胶因子的制备:
[0133][0134]
将l-n-phe的三氟乙酸盐溶于二氯甲烷,冰浴冷却,依次加入三乙胺、对苯二甲酰氯,将反应溶液缓慢升至室温并搅拌,室温反应24h。将所得胶体布氏漏斗抽滤,二氯甲烷、乙醇、去离子水依次洗涤,烘箱干燥样品,得到目标产物l-phe,对苯二甲酰氯与l-n-phe三氟乙酸盐和三乙胺的摩尔比为1:2.2:10。
[0135]
对比例2
[0136]
本对比例为d-phe的制备方法,步骤如下:
[0137]
步骤a1,boc-d-n-phe的制备
[0138]
boc-d-n-phe的制备:将boc-d-苯丙氨酸和1-羟基苯并三唑用二氯甲烷溶解,冰浴冷却,加入二甘醇胺和n,n-二异丙基乙胺,冰浴30min后,加入edci;将反应溶液缓慢升至室温,继续室温反应12h;依次用饱和柠檬酸溶液,饱和碳酸氢钠溶液,饱和氯化钠溶液洗涤,旋干,用乙酸乙酯和石油醚重结晶,其中boc-d-苯丙氨酸与二甘醇胺、n,n-二异丙基乙胺、1-羟基苯并三唑、edci的摩尔比为1:1.5:3.6:1.3:2。
[0139]
步骤a2,d-n-phe三氟乙酸盐的合成
[0140]
boc-d-n-phe加入到二氯甲烷/三氟乙酸(v/v)为4/1的溶液中,室温反应3h后,将溶液旋转蒸发完全除去溶液和剩余d-n-phe的三氟乙酸。
[0141]
步骤a3,氟原子不同取代位置凝胶因子的制备
[0142]
将d-n-phe的三氟乙酸盐溶于二氯甲烷,冰浴冷却,依次加入三乙胺、对苯二甲酰氯,将反应溶液缓慢升至室温并搅拌,室温反应24h。将所得胶体布氏漏斗抽滤,二氯甲烷、乙醇、去离子水依次洗涤,烘箱干燥样品,得到目标产物d-phe。
[0143]
本实施例制得的卤化效应功能化的超分子水凝胶,见图1a、1b和1c:图1a为氟化功能化的水凝胶,分别为l-2f-phe、l-3f-phe和l-4f-phe;图1b为不同卤素原子功能化的水凝胶,分别为l-4cl-phe、l-4br-phe、l-4i-phe、l-4f-es、l-4cl-es、l-4br-es、和l-4i-es;图1c为不同卤素原子功能化的水凝胶,分别为l-4f-pa、l-4cl-pa、l-4br-pa、和l-4i-pa。实施
例1制得的卤化效应功能化的手性结构的凝胶因子1h核磁谱图依次见图5-图12,图5为l-phe 1
hnmr谱,图6为l-2f-phe 1
h nmr谱,图7为l-3f-phe 1
h nmr谱,图8为l-4f-phe 1
hnmr谱,图9为l-4cl-phe 1
hnmr谱,图10为l-4br-phe 1
h nmr谱,图11为l-4i-phe 1
hnmr谱,图12为l-4f-es 1
h nmr谱,图13为l-4cl-es 1
hnmr谱,图14为l-4br-es 1
hnmr谱,图15为l-4i-es 1
hnmr谱,图16为l-4f-pa 1
h nmr谱,图17为l-4cl-pa 1
h nmr谱,图18为l-4br-pa 1
hnmr谱,图19为l-4i-pa 1
h nmr谱。
[0144]
使用扫描电镜(sem)对l-phe和氟化效应功能化的凝胶l-2f-phe、l-3f-phe和l-4f-phe微观形貌进行了表征分析。如图2所示,分别为a)l-phe、b)l-2f-phe、c)l-3f-phe、d)l-4f-phe,其中l-phe和l-2f-phe形成右手螺旋纤维,l-3f-phe和l-4f-phe形成左手螺旋纤维。
[0145]
进一步利用圆二色谱(cd)验证氟化效应诱导超分子手性的反转,如图4所示。图4a中,水凝胶l-phe的cd光谱显示出显著的科顿效应,在264nm处为零交叉点、303nm处偏弱的正效应峰和208nm处较强的负效应峰;类似地,图4b中,观察到水凝胶l-2f-phe的278nm处偏强的正效应峰和223nm处较弱的负效应峰;正如预期的那样,与凝胶l-phe和l-2f-phe相比,l-3f-phe和l-4f-phe的科顿效应在高波区发生了反转,l-3f-phe在229nm和315nm处具有两个负效应峰,l-4f-phe在230nm和300nm处具有两个负效应峰。结果与sem结果非常一致。
[0146]
应用例:氟化效应功能化的手性纳米纤维材料在细胞黏附、增殖方面的用途
[0147]
本发明中所用hepg2细胞和nih 3t3细胞由上海少辛生物科技有限公司代购。
[0148]
hepg2细胞和nih 3t3细胞在氟化效应功能化的手性均一的纳米纤维薄膜表面的黏附量调控
[0149]
本应用例采用的纳米纤维薄膜和三维水凝胶由前述实施例1制备的手性均一的苯丙氨酸衍生物凝胶因子自组装而得。
[0150]
对于二维纤维薄膜,首先将各种凝胶因子分别制备成0.3mg/ml超纯水稀溶液,冷却自组装,在96孔细胞培养板中每孔加入100μl 0.3mg/ml水凝胶稀溶液,37℃下真空干燥12h,紫外光照射30min消毒杀菌,将nih 3t3细胞和hepg2细胞接种在涂有功能化的干凝胶薄膜的96孔细胞培养板上(约2000个/孔),置于37℃,5%co2细胞培养箱中孵育,观察不同时间点细胞的黏附情况。采用活-死细胞双染,荧光显微镜下观察并拍照,用image j软件进行统计分析,见图3;由图3a和3b可知,nih 3t3细胞粘附在l-4f-phe(lm)纳米纤维膜上的数量是d-4f-phe(dp)膜上的2.2倍,是l-phe(lp)膜上的1.6倍,是d-phe(dm)膜上的1.3倍,是ps孔上的1.75倍。hepg2细胞粘附在l-4f-phe(lm)膜上的细胞数量分别是d-4f-phe(dp)膜上、l-phe(lp)膜上、d-phe(dm)膜上和ps孔上的细胞数量的1.9倍、1.3倍、1.1倍和1.5倍。
[0151]
对于三维水凝胶,首先将各种凝胶因子分别制备成dmso母液,再将nih 3t3细胞悬液细胞混合到dmso(最终dmso浓度:5%)的凝胶剂浓缩液中(最终凝胶剂浓度:10mg/ml),在几分钟内就形成了包裹细胞的水凝胶,然后向水凝胶中加入更多的dmem,并将其转移到标准培养条件(37℃,5%co2)的湿化培养箱中。培养3天后,细胞均匀分布在水凝胶中,不沉积(图3c)。从活细胞发出的绿色荧光可以看出,l-4f-phe(lm)水凝胶的细胞密度高于d-4f-phe(dp)、l-phe(lp)和d-phe(dm)水凝胶。
[0152]
综上所述,本实施制得的手性均一的纳米凝胶材料成功仿生出细胞外微环境的手性螺旋结构,分子手性和组装纤维手性都能够明显的影响细胞的粘附生长行为。研究证明,
高阶超分子手性对细胞增殖和活力的影响比单体手性的影响更显著,且超分子手性和单体手性对细胞增殖和活力的正相互作用顺序为lm》dm》lp》dp。
[0153]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1