一种瑞卢戈利的晶型及其制备方法与流程
1.本发明属于药物合成技术领域,具体地说是一种瑞卢戈利的晶型及其制备方法。
背景技术:
2.瑞卢戈利(relugolix)的化学名称是:n-[4-[1-[(2,6-二氟苯基)甲基]-5-[(二甲基氨基)甲基]-1,2,3,4-四氢-3-(6-甲氧基-3-哒嗪基)-2,4-二氧噻吩并[2,3-d]嘧啶-6-基]苯基]-n'-甲氧基脲,该化合物是有日本武田公司研发的促性腺激素释放激素拮抗剂(gnrh),于2018年在日本上市,用于子宫肌瘤引的出血和疼痛。
[0003]
其化学结构如示:
[0004][0005]
瑞卢戈利是一种小分子促性腺激素释放素(gnrh)受体拮抗剂,有潜力用于子宫纤维瘤、子宫内膜异位、前列腺癌等适应症。已经在临床试验的近1600名研究参与者中进行了评估。武田制药通过一系列在日本进行的临床iii期研究,对比了relugolix与leuprorelin(亮丙瑞林)治疗月经过多的子宫纤维化的安全性和有效性,以及上述两个药物在治疗与子宫纤维化有关的疼痛症状上的安全性和有效性,最终证实了relugolix用于子宫纤维瘤的安全性和有效性。此外,武田制药进行了relugolix的子宫内膜异位和前列腺癌的ii期临床研究,证实relugolix能显著减轻子宫内膜异位引起的疼痛,也能降低血清睾酮到去势水平并显著降低前列腺特异性抗原(psa)。随着销售额的不断上升,原料药瑞卢戈利的需求量也不断上升,因此对于原料药制备工艺的要求也越来越高,目前瑞卢戈利的制备工艺仍然存在路线长,成本高,环境不友好,质量不稳定等一系列问题。故对瑞卢戈利的制备工艺进行优化和改进具有确实的必要性。
[0006]
目前已有报道的瑞卢戈利的合成方法有多种,如:
[0007]
路线一(j.med.chem.2011,54,4998-5012):公开了的合成路线,该条路线为化合物路线,最后一步收率仅44%,收率偏低,合成路线如下:
[0008][0009]
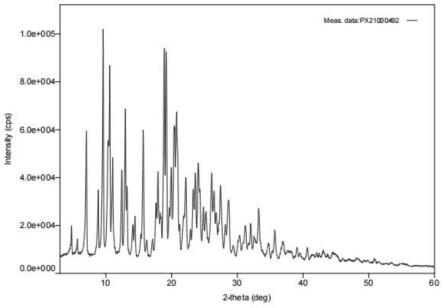
路线二:原研专利cn104703992a公开另外一条合成路线,原研瑞卢戈利x-射线粉末衍射测试谱图如图1所示,合成路线如下:
[0010][0011]
该条路线收率总体收率较高,具有工业化生产的可行性,但最后一步成脲反应,生成约2.0%的缩脲副产物,该杂质难以去除,此外该杂质随着投料规模的放大,杂质有增加的趋势,去除难度进一步加大,会影响药物最后的晶型,因此,开发一条收率高且缩脲副产物少的瑞卢戈利工艺路线将具有重要的意义。
技术实现要素:
[0012]
为了克服现有技术的缺点和不足,本发明提供了一种瑞卢戈利的晶型及其制备方法。
[0013]
本发明所采取的技术方案为:
[0014]
一种瑞卢戈利的晶型,其x射线粉末衍射图在2theta值为17.1、17.63、17.93、
18.32、18.87、19.19、19.64、19.94、20.43、20.72、20.91、21.22、21.81、22.16、22.86、23.28、23.61处具有特征峰。
[0015]
本发明还提供了上述瑞卢戈利晶型的制备方法,包括下述步骤:
[0016]
(1)称取化合物1,催化剂pt/c加入到氢化釜中,再加入溶剂a,将体系置换成氮气氛围,然后,将体系置换成氢气氛围,在室温下搅拌反应,过滤催化剂,用甲醇淋洗,滤液减压浓缩至干,减压干燥后得化合物2;
[0017][0018]
(2)将化合物2加入溶剂b中,搅拌溶清,控温搅拌下滴加入4m氯化氢二氧六环溶液,滴加完毕,保温搅拌,过滤,用乙腈洗涤,再用乙酸乙酯淋洗,再用正庚烷洗涤,减压烘干,得到化合物3;
[0019][0020]
(3)将乙腈和cdi加入反应瓶中,搅拌下加入三乙胺,将反应液冷却至5~15℃,保持温度10~30℃逐份加入盐酸甲氧基胺,加毕搅拌反应,反应液溶清;加入化合物3,升温至45~55℃搅拌反应后,再加入三乙胺;在40~55℃下向反应液滴加纯化水,搅拌析出晶体,过滤,干燥得化合物4;
[0021][0022]
(4)向反应瓶中加入化合物4、乙醇、水,搅拌下加入koh水溶液,搅拌升温至55~65℃,保温反应5~6h,反应完全后停止加热,搅拌降温至20~30℃,调节ph值,减压浓缩蒸除乙醇,加乙酸乙酯和乙醇打浆搅拌析晶,再加入水搅拌析晶,过滤,干燥,得化合物5;
[0023][0024]
(5)向反应瓶中搅拌下加入化合物5、哒嗪、dmac、dmap和、edc盐酸盐,升温至40~70℃保温搅拌,反应完毕后,降温至20~30℃,控制温度20~30℃加入水,析出固体,干燥得化合物6;
[0025][0026]
(6)向反应瓶中加入化合物6、甲醇、四氢呋喃和甲醇钠溶液,搅拌升温至50~80℃反应,反应完毕后,降温至室温,向里滴加浓盐酸,滴毕搅拌后,向里加入异丙醇,室温搅拌后降温搅拌,过滤,洗涤,干燥得化合物7;
[0027][0028]
(7)将化合物7加入dmso,升温至30~40℃搅拌溶清,再加入无水乙醇a,搅拌用滤纸过滤,用无水乙醇b洗涤滤饼,合并后搅拌保温至10~20℃,再加无水乙醇c,保温10~20℃搅拌12~16h,过滤,用乙醇洗涤,干燥得成品;其中化合物7、dmso、无水乙醇a、无水乙醇b、无水乙醇c的质量比为1:2.1~2.6:0.1~0.2:0.05~0.1:0.9~1.1。
[0029]
合成路线为:
[0030][0031]
作为优选,步骤(1)中,溶剂a为乙酸乙酯、甲苯、甲醇中的一种;步骤(2)中,溶剂b为二氧六环或乙酸乙酯。
[0032]
作为优选,步骤(1)具体为:称取化合物1 500克,催化剂pt/c 5克,加入到30l氢化釜中,再加入15l乙酸乙酯,将体系置换成氮气氛围,然后,将体系置换成氢气氛围,在室温下搅拌反应3小时,过滤催化剂,用甲醇淋洗,滤液减压浓缩至干,减压干燥后得化合物2,收率99.5%,化学纯度99.7%,其中脱氟杂质为0.002%。
[0033]
作为优选,步骤(2)具体为:将化合物2 100克加入0.5l二氧六环中,搅拌溶清,控温20℃搅拌下滴加入0.5l 4m氯化氢二氧六环溶液,滴加完毕,保温20℃搅拌3h,过滤,用乙腈洗涤,再用乙酸乙酯淋洗,再用正庚烷洗涤,50℃减压烘干,得到化合物3,收率为93%。
[0034]
作为优选,步骤(3)中具体为:将8l乙腈和1.06kg cdi加入反应瓶中,搅拌下加入367.6g三乙胺,将反应液冷却至10℃,保持温度10~20℃逐份加入580g盐酸甲氧基胺,加毕搅拌反应30min,反应液溶清;加入2kg化合物3,升温至50℃搅拌反应2h,反应完毕后,再加入415g三乙胺,搅拌10min;在40~50℃下向反应液滴加9kg纯化水,搅拌析出晶体,过滤,干燥得化合物4,收率为93%。
[0035]
作为优选,步骤(4)具体为:向反应瓶中加入5.6kg化合物4,50.4l乙醇,14.56l水,搅拌下加入2.16kg 48%koh水溶液,搅拌升温至60℃,保温反应5~6h,液相检测反应完全,停止加热,搅拌降温至25℃,用6m盐酸调节ph值为6~7,减压浓缩蒸除乙醇,向里加2.8l乙酸乙酯和2.8l乙醇打浆搅拌析晶,再加入12.6l水搅拌析晶,过滤,干燥,得化合物5,收率为93.7%。
[0036]
作为优选,步骤(5)具体为:向反应瓶中搅拌下加入3.85kg化合物5,1.023kg哒嗪,18.04kg dmac,0.53g dmap和1.85kg edc盐酸盐,升温至50~60℃保温搅拌1h,取样检测反应完毕;降温至25℃,控制温度20~30℃加入28.9kg水,析出固体,干燥,得化合物6,收率94.8%。
[0037]
作为优选,步骤(6)具体为:向反应瓶中加入4kg化合物6,79kg甲醇,6.02kg四氢呋喃和0.25kg 28%甲醇钠溶液,搅拌升温至60~70℃反应,液相检测反应完毕;降温至室温,向里滴加1.53kg浓盐酸,滴毕搅拌30min,向里加入31.4kg异丙醇,室温搅拌30min后降温0~10℃后搅拌1h,过滤,滤饼用5l冷甲醇/异丙醇=1/1洗涤,鼓风干燥,得化合物7,收率为98%。
[0038]
作为优选,步骤(7)具体为:将153g化合物7加入377g dmso,升温至35℃搅拌溶清,再加入27g无水乙醇,搅拌用滤纸过滤去除机械杂质,用11g无水乙醇洗涤滤饼,合并后搅拌保温至15℃,再向里加151g无水乙醇,保温15℃搅拌14h,过滤,用乙醇洗涤,50℃鼓风干燥得成品145g,收率为95%。
[0039]
与现有技术相比,本发明具有以下优势:
[0040]
(1)硝基还原能严格控制脱氟杂质的铂基催化体系,其中pt/c催化剂活化预处理后,在特定的溶剂中能催化氢化反应进行,反应转化率高、杂质少、氢化条件温和,而且催化剂可以套用10次以上,脱氟杂质都能得到较好的控制(小于0.01%);
[0041]
(2)该路线采用氨化物2成盐具有明显优点:氢化后的氨基物2为油状物,含有一定的残留溶剂,难以确定下一步的投料比例,对后续成脲反应有较大影响,要么反应不完要么会产生上两个脲分子杂质,对后续中间体杂质产生传递,影响最终产物的纯度,从而影响其晶型;
[0042]
(3)通过上述关键步骤实验条件的控制,通过高纯度的中间体来制备新晶型的原料药,该原料药经过结构表征,未见文献报道。
附图说明
[0043]
图1是原研瑞卢戈利x-射线粉末衍射测试谱图;
[0044]
图2是瑞卢戈利新晶型x-射线粉末衍射测试谱图。
具体实施方式
[0045]
下面通过实施例,对本发明的技术方案作进一步具体的说明,这些实施例是对本发明的说明而作,不是对本发明的限制。基于本技术中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本技术保护的范围。
[0046]
本发明中涉及的原料如没有特殊说明,均来源于市场购买。
[0047]
实施例1
[0048]
制备化合物2
[0049][0050]
称取化合物1 500克,催化剂pt/c 5克加入到30l氢化釜中,再加入15l乙酸乙酯,将体系置换成氮气氛围,然后,将体系置换成氢气氛围,在室温下搅拌反应3小时,过滤催化剂,用甲醇淋洗,滤液减压浓缩至干,减压干燥后得化合物2,收率99.5%,化学纯度99.7%,其中脱氟杂质为0.002%。
[0051]1h nmr(500mhz,dmso
d6
)δ8.43(d,j=8.0hz,1h),8.29(s,1h),8.27(d,j=5.3hz,1h),7.78(s,1h),7.51(d,j=6.5hz,2h),7.27
–
7.23(m,1h),7.19
–
7.14(m,2h),6.76(s,1h),4.58(s,2h),3.88(s,3h),3.74(s,3h),2.89(t,j=6.7hz,2h),2.63(s,3h),2.36(t,j=6.7hz,2h),2.17(s,6h).
[0052]
制备化合物3
[0053][0054]
将上述步骤中的化合物2 100克加入0.5l二氧六环搅拌溶清,控温20℃搅拌下滴加入0.5l 4m氯化氢二氧六环溶液,随着滴加一半后有大量晶体析出,滴加完毕,保温20℃搅拌3h,过滤,用少量乙腈洗涤,再用乙酸乙酯淋洗,再用正庚烷洗涤,50℃减压烘干,得到类白色固体(化合物3),易吸湿,快速装袋称重为100g,收率为93%。
[0055]
化合物4的制备
[0056]
将8l乙腈和1.06kg cdi加入反应瓶中,搅拌下加入367.6g三乙胺,将反应液冷却至10℃,保持温度10~20℃逐份加入580g盐酸甲氧基胺,加毕搅拌反应30min,反应液溶清。加入2kg化合物3,升温至50℃搅拌反应2h,取样检测,再加入415g三乙胺,搅拌10min。在40~50℃下向反应液缓慢滴加9kg纯化水,搅拌会析出晶体,过滤,干燥得化合物4,收率为93%。
[0057]
化合物5的制备
[0058]
向反应瓶中加入5.6kg化合物4,50.4l乙醇,14.56l水,搅拌下加入2.16kg48%koh水溶液,搅拌升温至60℃,保温反应5~6h,液相检测反应完全,停止加热,搅拌降温至25℃,用6m盐酸调节ph值为6~7,减压浓缩蒸除乙醇,向里2.8l乙酸乙酯和2.8l乙醇打浆搅拌析晶,再加入12.6l水搅拌析晶,过滤,干燥,得化合物5,收率为93.7%。
[0059]
化合物6的制备
[0060]
向反应瓶中搅拌下加入3.85kg化合物5,1.023kg哒嗪,18.04kgdmac,0.53gdmap和
1.85kgedc盐酸盐,升温至50~60℃保温搅拌1h,(反应液先澄清后析出固体),取样检测反应完毕。降温至25℃,控制温度20~30℃加入28.9kg水,析出大量固体,干燥,收率94.8%。
[0061]
化合物7的制备
[0062]
向反应瓶中加入4kg化合物6,79kg甲醇,6.02kg四氢呋喃和0.25kg28%甲醇钠溶液,搅拌升温至60~70℃反应,液相检测反应完毕。降温至室温向里滴加1.53kg浓盐酸,滴毕搅拌30min,向里加入31.4kg异丙醇,室温搅拌30min后降温0~10℃后搅拌1h,过滤,滤饼用5l冷甲醇/异丙醇=1/1洗涤,鼓风干燥得,收率为98%。
[0063]
瑞卢戈利新晶型制备方法
[0064]
将153g化合物7加入377g dmso,升温至35℃搅拌溶清,再加入27g无水乙醇,搅拌用滤纸过滤去除机械杂质,用11g无水乙醇洗涤滤饼,合并后搅拌保温至15℃,再向里缓慢流加151g无水乙醇,保温15℃搅拌14h,过滤,用乙醇洗涤,50℃鼓风干燥得成品145g,收率为95%。瑞卢戈利新晶型的x-射线粉末衍射测试谱图如图2所示。
[0065]
本发明xrd 2θ值与原研公司2θ值对比见表1。
[0066]
表1
[0067]
[0068]
[0069]
[0070][0071]
实施例2
[0072]
称取化合物1 500克,催化剂pt/c 5克加入到30l氢化釜中,再加入15l甲苯,将体系置换成氮气氛围,然后,最后将体系置换成氢气氛围,在室温下搅拌反应3小时,过滤催化剂,用甲醇淋洗,滤液减压浓缩至干,减压干燥后得化合物2,收率99.2%,化学纯度99.2%,其中脱氟杂质为0.003%。
[0073]
实施例3
[0074]
称取化合物1 500克,催化剂pt/c 5克加入到30l氢化釜中,再加入15l甲醇,将体系置换成氮气氛围,然后,最后将体系置换成氢气氛围,在室温下搅拌反应3小时,过滤催化剂,用甲醇淋洗,滤液减压浓缩至干,减压干燥后得化合物2,收率99.1%,化学纯度99.3%,其中脱氟杂质为0.005%。
[0075]
实施例4
[0076]
将实施例1中的化合物2 100克加入0.5l乙酸乙酯搅拌溶清,控温20℃搅拌下滴加入0.5l 4m氯化氢乙酸乙酯溶液,随着滴加一半后有大量晶体析出,滴加完毕,保温20℃搅拌3h,过滤,用少量二氧六环洗涤,再用乙酸乙酯淋洗,再用正庚烷洗涤,50℃减压烘干,得到类白色固体,易吸湿,快速装袋称重为97g,收率为90%。
[0077]
实施例5
[0078]
称取化合物1 500克,第10次套用的催化剂pt/c 5克加入到30l氢化釜中,再加入15l甲苯,将体系置换成氮气氛围,然后,最后将体系置换成氢气氛围,在室温下搅拌反应3小时,过滤催化剂,用甲醇淋洗,滤液减压浓缩至干,减压干燥后得化合物2,收率99.0%,化学纯度99.2%,其中脱氟杂质为0.004%。
[0079]
以上各实施例仅用以说明本技术的技术方案,而非对其限制;尽管参照前述各实施例对本技术进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本技术各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1