一种提高基因表达水平的酿酒酵母工程菌及其构建方法与应用
1.本发明属于生物技术领域,具体涉及一种提高基因表达水平的酿酒酵母工程菌及其构建方法与应用。
背景技术:
2.酿酒酵母(saccharomyces cerevisiae),又称面包酵母,长期应用于酿酒、面包和馒头制作等,安全可靠,不产生毒素,是国际公认的食品安全级(generally regarded as safe,gras)真核微生物;由于其菌体细胞富含营养,具很高的经济价值,酵母抽提物或酵母浸出物(yeast extract)不仅广泛用于微生物、动植物细胞培养,在制药、酿造及发酵食品中有着举足轻重的作用,而且也直接用于饲料及食品添加剂;由于酵母在工业生产中具有良好的发酵性能,在发酵过程中能够快速分裂,容易培养,对杂菌污染具有较强的抗性,遗传背景清晰,基因操作简单等优势,使其在基因工程技术中常被用于代谢工程改造的出发菌株;在新近推崇的合成生物学研究中,酿酒酵母因其特殊的代谢能力等特点已成为备受关注的底盘菌株,常用来合成异源高附加值产物。但异源代谢产物途径相关基因的组成型表达通常会导致细胞资源的过多消耗,抑制细胞生长,从而影响了目标产物的最终产量。
3.为将细胞生长与产物合成解偶联,同时实现目标产物合成基因的高效表达,目前常用的诱导调控的方法如半乳糖诱导型启动子诱导异源途径基因表达,在细胞浓度达到一定值后添加诱导剂半乳糖,从而避免细胞生长初期过多的资源消耗。但如半乳糖等诱导物价格十分昂贵,工业化生产成本较高。
4.酿酒酵母具有内源性的与细胞交配相关的信息素介导的转录调控系统,因此还可以以酿酒酵母内源性信息素响应途径为基础构建基因表达调控体系,实现外源基因的表达。但是使用该系统表达外源基因时,外源基因的表达水平会低于半乳糖代谢调控系统启动子p
gal1
所驱动的基因表达水平,不适用于异源代谢产物合成。并且基于酿酒酵母内源性信息素sα的群体感系统感应α因子的浓度阈值较低,在激活时会将细胞周期阻滞在g1期,严重抑制细胞生长,不利于制备异源代谢产物。因此进一步构建一种提高基因表达水平的酿酒酵母工程菌是十分必要的。
技术实现要素:
5.针对现有技术的不足,本发明提供了一种提高基因表达水平的酿酒酵母工程菌及其构建方法与应用。
6.本发明的技术方案如下:
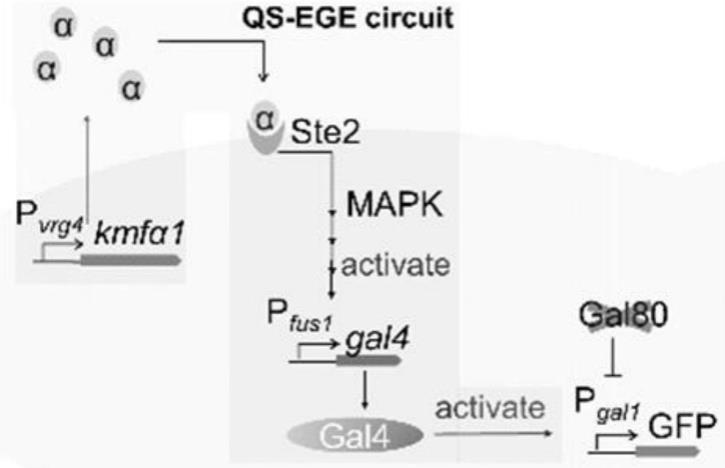
7.一种提高基因表达水平的酿酒酵母工程菌,是以酿酒酵母w303-1a为出发菌株,该酿酒酵母工程菌敲除了胞外蛋白酶编码基因bar1和半乳糖代谢途径抑制因子编码基因gal80,同时含有以启动子p
vrg4
表达的乳酸克鲁维酵母来源的kα因子编码基因kmfα1和以启动子p
fus1
表达的转录激活因子编码基因gal4。
8.上述酿酒酵母工程菌的构建方法,包括步骤:以酿酒酵母w303-1a为出发菌株,敲除bar1基因,然后转化含有启动子p
vrg4
和kα因子编码基因kmfα1的重组载体prs304-p
vrg4-kmfα1-t
cyc1
,将gal4编码基因的原始启动子替换为p
fus1
,再转化融合片段p
fus1-gal4,最后敲除gal80基因,经筛选、验证获得。
9.根据本发明优选的,所述提高基因表达水平的酿酒酵母工程菌的构建方法,具体包括步骤如下:
10.(1)以质粒pumri-a中两端带有loxp序列(cre酶切割位点)的kanmx遗传霉素标记基因为模板,以bar1-g418-up-f和bar1-g418-down-r为引物进行pcr扩增,得到bar1基因的敲除组件;然后将bar1基因的敲除组件转化至酿酒酵母菌株w303-1a,经筛选后得到敲除胞外蛋白酶bar1编码基因的菌株bar1δ;
11.(2)人工合成密码子优化后的α因子编码基因kmfα1序列,然后进行pcr扩增,得到优化后的kmfα1序列;以酿酒酵母w303-1a基因组dna为模板进行pcr扩增,得到启动子p
vrg4
和终止子t
cyc1
;将p
vrg4
、优化后的kmfα1序列和t
cyc1
进行融合pcr,再将融合pcr产物p
vrg4-kmfα1-t
cyc1
连接到质粒prs304上,得到重组载体prs304-p
vrg4-kmfα1-t
cyc1
;将重组载体prs304-p
vrg4-kmfα1-t
cyc1
转化至菌株bar1δ中,经筛选后得菌株vrg4p-kα;
12.(3)以酿酒酵母w303-1a基因组dna为模板进行pcr扩增,得到带有启动子p
gal4
下游同源臂序列的启动子p
fus1
,以pumri-a质粒为模板进行pcr扩增,得到带有p
gal4
上游同源臂序列的kanmx编码序列;将启动子p
fus1
和kanmx序列进行融合pcr扩增,得融合pcr产物p
fus1-gal4片段;将p
fus1-gal4片段转化至菌株vrg4p-kα,经筛选后得菌株kα-ege1;
13.(4)以质粒pumri-a的kanmx遗传霉素标记基因为模板,以gal80-knockout-f/r为引物进行pcr扩增,,得到gal80基因的敲除组件;然后将得到的gal80基因敲除组件转化至酿酒酵母菌株kα-ege1,经筛选后得到提高基因表达水平的酿酒酵母工程菌kα-ege2。
14.根据本发明优选的,步骤(1)中,所述bar1-g418-up-f带有bar1上游50bp同源臂,bar1-g418-down-r带有bar1下游50bp同源臂,具体序列如下:
15.bar1-g418-up-f:5
′‑
taacatgtatacacagccagctattctgaaacacaccacattatagataacttcgtataatgtatgc-3
′
,
16.bar1-g418-down-r:5
′‑
ataatgtgctacttgttcaaaattgtgatggctgcataatattacataacttcgtatagcatac-3
′
。
17.根据本发明优选的,步骤(1)中,所述loxp的序列如下:
18.loxp:5'-ataacttcgtatagcatacattatacgaagttat-3'。
19.根据本发明优选的,步骤(2)中,所述α因子来自于克鲁维酵母,核苷酸序列如seq id no.1所示,pcr扩增引物序列如下:
20.kmfa1-f:5
′‑
atgaaattctctactatattag-3
′
,
21.kmfa1-r:5
′‑
attacatgatcagaaaattggttggcc-3
′
。
22.根据本发明优选的,步骤(2)中,所述启动子p
vrg4
的pcr扩增引物序列如下:
23.304-bamhi-vrg4p-f:5
′‑
cgctctagaactagtggatcccaaacaacaatttcaacag-3
′
,
24.vrg4p-mfa1-r:5
′‑
tatagtagagaatttcattcgggcgaaagatactg-3
′
;
25.所述终止子t
cyc1
的pcr引物序列如下:
26.cyc1t-kmfa1-f:5
′‑
caattttctgatcatgtaattagttatg-3
′
;
27.304-xhoi-cyc1t-r:5
′‑
gtaccgggccccccctcgaggcaaattaaagccttcg-3
′
。
28.根据本发明优选的,步骤(3)中,所述带有启动子p
gal4
下游同源臂的启动子p
fus1
的pcr引物序列如下:
29.fus1-f:5
′‑
atcaacaacagggtcagc-3
′
;
30.fus1-down-pgal4-r:5
′‑
ttaagtcggcaaatatcgcatgcttgttcgatagaagacagtagcttcattttgattttcagaaacttgatg-3
′
;
31.所述带有启动子p
gal4
上游同源臂的kanmx遗传霉素标记基因的pcr扩增引物序列如下:
32.up-pgal4-g418-f:5
′‑
tcaaagtatttacataattctgtatcagtttaatcaccataatatcgtttataacttcgtataatgtatg-3
′
,
33.g418-fus-r:5
′‑
gctgaccctgttgttgatataacttcgtatagc-3
′
。
34.根据本发明优选的,步骤(3)中,所述融合pcr中启动子p
fus1
和转录激活因子gal4的摩尔比为1:1。
35.根据本发明优选的,步骤(4)中,所述gal80-knockout-f带有gal80上游50bp同源臂,gal80-knockout-r带有gal80下游50bp同源臂,具体序列如下:
36.gal80-knockout-f:5
′‑
gtatacaatctcgatagttggtttcccgttctttccactcccgtctaacttcgtataatgtatgc-3
′
;
37.gal80-knockout-r:5
′‑
ttacccacaatggcattataatttcgtaaatgatatacttccatgataacttcgtatagcatac-3
′
。
38.上述提高基因表达水平的酿酒酵母工程菌在构建表达外源基因的基因工程菌中的应用。
39.根据本发明优选的,所述表达外源基因的基因工程菌为含有外源基因或含该外源基因的载体的菌株,所述菌株的基因组中整合了所述外源基因。
40.根据本发明优选的,所述外源基因为工业、饲料或食品领域中用到的蛋白的编码序列。
41.进一步优选的,所述外源基因为酶的编码序列,所述酶为糖基转移酶。
42.最优选的,所述外源基因为α-1,3-岩藻糖基转移酶基因或α-1,2-岩藻糖基转移酶基因。
43.本发明的技术特点:
44.本发明将启动子p
vrg4
、α因子kmfα1、启动子p
fus1
和转录激活因子gal4构成qs-ege2系统,然后将该系统构建在敲除了胞外蛋白酶编码基因bar1和转录抑制因子编码基因gal80的酿酒酵母中。其中,在酿酒酵母中以弱启动子p
vrg4
表达克鲁维酵母来源的kα因子,明显降低了酿酒酵母α信息素调控系统激活时对细胞生长造成的抑制,利用这一激活的系统中转录水平上调较高的p
fus1
驱动半乳糖代谢途径转录激活因子gal4的表达,进而利用gal4进一步激活启动子p
gal
所表达的目标基因,取得了可明显提高基因表达水平的效果。敲除bar1可防止分泌至胞外的α因子被降解,而敲除gal80可以解除半乳糖代谢途径的抑制作用,进一步放大该系统的输出水平。
45.本发明的有益效果:
46.1、本发明以酿酒酵母内源性启动子p
fus1
和转录激活因子gal4序列构成的信息素
感应系统为基础,通过异源信息素α因子kmfα1的表达结合转录信号放大,得到了由启动子p
vrg4
、α因子kmfα1、启动子p
fus1
和转录激活因子gal4构成的qs-ege2系统。该系统可以实现外源基因的高水平表达。并且在外源基因高效表达的基础上,避免了酿酒酵母内源性α因子对细胞生长的抑制。
47.2、本发明提供的提高基因表达水平的酿酒酵母工程菌中含有基因表达调控系统(qs-ege2系统),可用于调控包括糖基转移酶在内的异源产物途径相关酶,可通过增强其表达水平进而取得提高目标产物合成效率的效果。与酿酒酵母合成异源产物时常用的半乳糖诱导型启动子相比,本发明提供的qs-ege2系统不需要额外添加半乳糖诱导基因的表达,在成本低廉的葡萄糖碳源下即可实现基因的高效表达,且基因的表达水平为半乳糖诱导型启动子p
gal1
的2.7倍。利用本发明提供的提高基因表达水平的酿酒酵母工程菌进行2-岩藻糖基乳糖和3-岩藻糖基乳糖的合成时,明显提高了2-岩藻糖基乳糖和3-岩藻糖基乳糖的合成效率,证实了本发明的工程菌和qs-ege2系统具有良好的实际应用价值。
附图说明
48.图1为菌株kα-ege2的构建示意图。
49.图2为工程菌kα-fus1p-gfp中基因表达调控系统强度图。
50.图3为工程菌kα-ege2-gfp中qs-ege2系统强度图。
51.图中:左图为kα-ege2-gfp和仅以半乳糖诱导型启动子表达gfp的菌株gal-gfp分别在葡萄糖碳源和半乳糖碳源下的生长曲线,右图为kα-ege2-gfp和半乳糖诱导下的菌株gal-gfp中的gfp表达强度。
52.图4为菌株kα-ege3-gfp菌株构建示意图。
53.图5为工程菌ege3-gfp中qs-ege3系统强度图。
54.图中:左图为葡萄糖培养下的kα-ege2-gfp和kα-ege3-gfp生长曲线,右图葡萄糖培养下的kα-ege2-gfp和kα-ege3-gfp中gfp表达强度。
55.图6为菌株kα-ege4-2fl和对照菌株fl06发酵产物中的2
’‑
fl产量图。
56.图中:左图为葡萄糖培养下的kα-ege4-2fl和半乳糖培养下的对照菌株fl06生长曲线,右图为葡萄糖培养下的kα-ege4-2fl和半乳糖培养下的对照菌株fl06中2
’‑
fl产量。
57.图7为菌株kα-ege4-2fl和对照菌株fl303发酵产物中的3
’‑
fl产量图。
58.图中:左图为葡萄糖培养下的kα-ege4-3fl和半乳糖培养下的对照菌株fl303生长曲线,右图为葡萄糖培养下的kα-ege4-3fl和半乳糖培养下的对照菌株fl303中3-fl产量。
具体实施方式
59.下面结合实施例和附图对本发明的技术方案作进一步说明,但是本发明的保护范围并不仅限于此。实施例中涉及的试剂及药品,若无特殊说明,均为普通市售产品;实施例中涉及的实验操作,若无特殊说明,均为本领域常规操作。
60.将克鲁维酵母来源的α因子编码基因进行酿酒酵母密码子优化后,在金唯智公司合成相应编码基因序列,得到α因子kmfα1序列;
61.本发明中使用的酿酒酵母w303-1a为普通市售菌种,可从微生物保藏中心或菌种销售公司购得。
62.实施例1:胞外蛋白酶bar1编码基因的敲除
63.1、以质粒pumri-a中两端带有loxp序列的kanmx遗传霉素标记基因为模板,以带有bar1上游50bp同源臂的bar1-g418-up-f和带有bar1下游50bp同源臂的bar1-g418-down-r为引物进行pcr扩增,得到bar1基因的敲除组件(seq id no.6),所述引物具体序列如下:
64.bar1-g418-up-f:5
′‑
taacatgtatacacagccagctattctgaaacacaccacattatagataacttcgtataatgtatgc-3
′
,
65.bar1-g418-down-r:5
′‑
ataatgtgctacttgttcaaaattgtgatggctgcataatattacataacttcgtatagcatac-3
′
;
66.pcr扩增体系:使用购自vazyme的高保真dna聚合酶phanta super-fidelity dnapolymerase,pcr扩增体系按照该试剂盒说明书配制。
67.pcr扩增程序:预变性95℃3min,变性95℃15s,复性55℃15s,延伸72℃1min/kb,30个循环,后延伸72℃5min,12℃保存。
68.2、将bar1基因的敲除组件转化至酿酒酵母菌株w303-1a,在带有200μg/ml的g418抗生素的ypd固体培养基上筛选阳性转化子后,得敲除胞外蛋白酶bar1编码基因的菌株bar1δ。
69.所述ypd固体培养基的成分为:20g/l葡萄糖,20g/l蛋白胨,10g/l酵母提取物。
70.实施例2:工程菌kα-ege1的构建
71.1、将克鲁维酵母来源的α因子编码基因kmfα1序列进行酿酒酵母密码子优化,然后由苏州金唯智生物科技有限公司合成相应编码基因序列,以该序列为模板进行pcr扩增,得到优化后的kmfα1序列(seq id no.1),pcr扩增引物序列如下:
72.kmfa1-f:5
′‑
atgaaattctctactatattag-3
′
,
73.kmfa1-r:5
′‑
attacatgatcagaaaattggttggcc-3
′
。
74.以酿酒酵母w303-1a基因组dna为模板进行pcr扩增,得到启动子p
vrg4
和终止子t
cyc1
,pcr扩增引物序列如下:
75.304-bamhi-vrg4p-f:5
′‑
cgctctagaactagtggatcccaaacaacaatttcaacag-3
′
,
76.vrg4p-mfa1-r:5
′‑
tatagtagagaatttcattcgggcgaaagatactg-3
′
;
77.cyc1t-kmfa1-f:5
′‑
caattttctgatcatgtaattagttatg-3
′
;
78.304-xhoi-cyc1t-r:5
′‑
gtaccgggccccccctcgaggcaaattaaagccttcg-3
′
。
79.将p
vrg4
、优化后的kmfα1序列和t
cyc1
按照1:3:1的摩尔比进行融合pcr,得融合pcr产物p
vrg4-kmfα1-t
cyc1
;将p
vrg4-kmfα1-t
cyc1
通过t5外切酶连接的方法连接到质粒prs304的xhoi和bamhi位点之间,得到重组载体prs304-p
vrg4-kmfα1-t
cyc1
;将重组载体prs304-p
vrg4-kmfα1-t
cyc1
以内切酶bsu36i线性化后,转化至菌株bar1δ中,通过色氨酸缺陷的sc固体培养基筛选,得菌株vrg4p-kα。
80.所述融合pcr采用vazymephanta super-fidelity dna polymerase试剂盒完成,融合pcr体系按照该试剂盒说明书配制。
81.所述启动子p
vrg4
的核苷酸序列如seq id no.3所示,所述终止子t
cyc1
的核苷酸序列如seq id no.5所示。
82.2、以酿酒酵母w303-1a基因组dna为模板,扩增得到带有启动子p
gal4
下游同源臂的启动子p
fus1
片段;以质粒pumri-a为模板,扩增得到带有启动子p
gal4
上游同源臂的kanmx遗传
霉素标记基因,pcr引物序列如下:
83.fus1-f:5
′‑
atcaacaacagggtcagc-3
′
;
84.fus1-down-pgal4-r:5
′‑
ttaagtcggcaaatatcgcatgcttgttcgatagaagacagtagcttcattttgattttcagaaacttgatg-3
′
;
85.up-pgal4-g418-f:5
′‑
tcaaagtatttacataattctgtatcagtttaatcaccataatatcgtttataacttcgtataatgtatg-3
′
,
86.g418-fus-r:5
′‑
gctgaccctgttgttgatataacttcgtatagc-3
′
。
87.将带有cre重组酶的载体yep-ch转化至工程菌vrg4p-kα中,在带有200μg/ml的hygromycin抗生素的ypd固体培养基上进行筛选,所得到的转化子通过半乳糖诱导cre重组酶表达,切除loxp位点之间的kanmx遗传霉素标记基因。将启动子p
fus1
和kanmx序列按照1:1的摩尔比进行融合pcr,得融合pcr产物p
fus1-gal4片段;将p
fus1-gal4片段转化至菌株vrg4p-kα,在带有200μg/ml的g418抗生素的ypd固体培养基上筛选阳性转化子后,得到菌株kα-ege1。
88.所述启动子p
fus1
的核苷酸序列如seq id no.4所示;所述p
fus1-gal4片段的核苷酸序列如seq id no.7所示
89.所述带有cre重组酶的载体的构建方法可参考文献:li,h.,shen,y.,wu,m.,hou,j.,jiao,c.,li,z.,liu,x.,and bao,x.(2016)engineering a wild-type diploid saccharomyces cerevisiaestrain for second-generation bioethanol production,bioresources and bioprocessing 3,51。
90.本步骤所得的工程菌kα-ege1中含有由启动子p
vrg4
、α因子kmfα1和元件p
fus1-gal4构成的qs-ege1系统。
91.3、以pumri-a质粒为模板,以带有gal80上游50bp同源臂的gal80-knockout-f和带有gal80下游50bp同源臂的gal80-knockout-r为引物进行pcr扩增,得到gal80基因的敲除组件(seq id no.8),pcr扩增引物序列如下:
92.gal80-knockout-f:5
′‑
gtatacaatctcgatagttggtttcccgttctttccactcccgtctaacttcgtataatgtatgc-3
′
;
93.gal80-knockout-r:5
′‑
ttacccacaatggcattataatttcgtaaatgatatacttccatgataacttcgtatagcatac-3
′
。
94.将带有cre重组酶的载体yep-ch转化至工程菌kα-ege1中,在带有200μg/ml的hygromycin抗生素的ypd固体培养基上进行筛选,所得到的转化子通过半乳糖诱导cre重组酶表达,切除loxp位点之间的kanmx遗传霉素标记基因,然后将gal80基因的敲除组件转化至该菌株中,在带有200μg/ml的g418抗生素的平板上筛选阳性转化子,得到提高基因表达水平的酿酒酵母工程菌kα-ege2。
95.菌株kα-ege2的构建示意图如图1所示,本实施例构建的工程菌kα-ege2中含有由启动子p
vrg4
、α因子kmfα1、启动子p
fus1
和转录激活因子gal4构成的qs-ege-2系统。
96.本实施例中所述pcr扩增体系、pcr扩增程序、融合pcr体系和融合pcr程序同实施例1。
97.实施例3:α因子提高gfp基因表达水平的验证
98.1、由苏州金唯智生物科技有限公司合成gfp基因(genbank登录号为cak02784.1),
以gfp基因为模板进行pcr扩增,pcr扩增引物序列如下:
99.yegfp-pfus1-f:5
′‑
ctgaaaatcaaaatgtctaaaggtgaag-3
′
;
100.yegfp-r:5
′‑
aattacatgattatttgtacaattcatc-3
′
。
101.以酿酒酵母w303-1a基因组dna为模板进行pcr扩增,得到启动子p
fus1
和终止子t
cyc1
,pcr扩增引物序列如下:
102.305-xbai-pfus1-f:5
′‑
caccgcggtggcggccgctctagaatcaacaacagggtc-3
′
;
103.pfus1-yegfp-r:5
′‑
tttagacattttgattttcagaaacttg-3
′
;
104.cyc1t-egfp-1-f:5
′‑
caaataatcatgtaattagttatg-3
′
,
105.305-hindiii-cyc1t-1-r:5
′‑
106.ggtcgacggtatcgataagcttcttcgagcgtcccaaaac-3
′
。
107.将p
fus1
、gfp基因和t
cyc1
按照1:3:1的摩尔比进行融合pcr,得融合pcr产物p
vrg4-kmfα1-t
cyc1
;将p
fus1-gfp-t
cyc1
连接到质粒prs305的hindiii和xbai酶切位点之间,得到重组载体prs305-p
fus1-gfp-t
cyc1
;将重组载体prs305-p
fus1-gfp-t
cyc1
以内切酶bspti线性化后,转化至菌株vrg4p-kα中,通过亮氨酸缺陷的sc固体培养基筛选,得菌株kα-fus1p-gfp。
108.2、对菌株kα-fus1p-gfp进行系统输出强度测试,结果如图2所示。
109.具体方法为:将待测菌株等量转接至ypd培养基中,摇床中200rpm,30℃培养。检测时取200μl菌液,以ddh2o清洗一次后转移至96孔板中,以荧光酶标仪(perkinelmer,1420multilabel counter)检测gfp强度,检测时设置激发光485nm,吸收光535nm,检测时长1s,读数为gfp强度。随后将样品在普通酶标仪中检测od 600吸收,读数可表征细胞生长量。
110.由图2可知,在引入α因子后的工程菌kα-fus1p-gfp中外源基因gfp的表达强度明显提高。
111.实施例4:gfp基因在工程菌kα-ege2中的表达
112.1、按照实施例3中的方法获得gfp基因。
113.以酿酒酵母w303-1a基因组dna为模板进行pcr扩增,得到启动子p
gal1
和终止子t
cyc1
,pcr扩增引物序列如下:
114.305-bamhi-pgal1-f:5
′‑
ccgctctagaactagtggatcccggattagaagccgccg-3
′
;
115.pgal1-yegfp-r:5
′‑
ttcacctttagacataatattccctatag-3
′
;
116.cyc1t-egfp-2-f:5
′‑
aattgtacaaataaccggtcttgctagattc-3
′
,
117.305-hindiii-cyc1t-2-r:5
′‑
118.ggtcgacggtatcgataagcttcttcgagcgtcccaaaac-3
′
。
119.将p
gal1
、gfp基因和t
cyc1
按照1:3:1的摩尔比进行融合pcr,得融合pcr产物p
gal1-gfp-t
cyc1
;将p
gal1-gfp-t
cyc1
连接到质粒prs305的bamhi和hindiii酶切位点之间,得到重组载体prs305-p
gal1-gfp-t
cyc1
;将重组载体prs305-p
gal1-gfp-t
cyc1
以内切酶bspti线性化后,分别kα-ege2中,通过亮氨酸缺陷的sc固体培养基筛选,得工程菌kα-ege2-gfp。所述启动子p
gal1
的核苷酸序列如seq id no.9所示。
120.将质粒prs305-p
gal1-gfp-t
cyc1
直接转化至酿酒酵母w303-1a中得到对照工程菌gal-gfp。
121.2、对表达外源基因gfp的工程菌kα-ege2-gfp进行qs-ege2系统输出强度测试,结果如图3所示。
122.具体方法为:将等量的工程菌gal-gfp和工程菌kα-ege2-gfp分别转接至ypd培养基中,摇床中200rpm,30℃培养。检测时取200μl菌液,以ddh2o清洗一次后转移至96孔板中,以荧光酶标仪(perkinelmer,1420multilabel counter)检测gfp强度,检测时设置激发光485nm,吸收光535nm,检测时长1s,读数为gfp强度。随后将样品在普通酶标仪(tecan 2000)中检测od 600
吸收,读数可表征细胞生长量。
123.由图3可知,含有由启动子p
vrg4
、α因子kmfα1、启动子p
fus1
和转录激活因子gal4构成的qs-ege2系统的工程菌kα-ege2-gfp对gfp的表达水平是gal-gfp激活状态下启动子p
gal1
的2.7倍。
124.实施例5:α因子的起始诱导作用对qs-ege2系统输出的影响
125.1、以质粒prs304为模板进行pcr扩增,分别得到kα因子表达盒上下游同源臂,以质粒pumri-a为模板扩增kanmx遗传霉素标记基因,通过pcr融合,将以上三个片段连接,得到α因子敲除表达盒,pcr扩增引物序列如下:
126.up-vk-f:5
′‑
ttctgaagatagaacgcatttttg-3
′
;
127.up-vk-r:5
′‑
cattacgcgtttaggcg-3
′
;
128.g418-vk-f:5
′‑
gaaaaatatcacagttgacgaaagaagacacgtcgcctaaacgcgtaatgataacttcgtataatgtatgctatacg-3
′
;
129.g418-vk-r:5
′‑
gcaaattaaagccttcgagcgtcccaaaaccttctcaagcaaggttttcattgatataacttcgtatagcatacattatac-3
′
;
130.up-vk-f:5
′‑
gttatatcaactagtgcttggagttgg-3
′
;
131.up-vk-r:5
′‑
gcaaattaaagccttcgagc-3
′
。
132.将带有cre重组酶的载体pyep-ch转化至工程菌kα-ege2-gfp中,在带有200μg/ml的hygromycin抗生素的ypd固体培养基上进行筛选,所得到的转化子通过半乳糖诱导cre重组酶表达,切除loxp位点之间的kanmx遗传霉素标记基因,再向该转化子中转化α因子敲除表达盒,在带有200μg/ml的g418抗生素的平板上筛选阳性转化子,得工程菌ege3-gfp。工程菌ege3-gfp中的基因表达调控系统命名为qs-ege3。菌株ege3-gfp的构建示意图如图4所示。
133.4、对工程菌ege3-gfp进行qs-ege3系统输出强度测试,结果如图5所示,测试方法同实施例4。
134.由图5可知,没有α因子kmfα1的qs-ege3系统对gfp的表达水平仅为工程菌kα-ege2-gfp的qs-ege2系统的26%,说明α因子kmfα1序列在qs-ege2系统中起到了提高外源基因表达水平中的重要作用。
135.实施例6:利用工程菌kα-ege2制备2-岩藻糖基乳糖(2
’‑
fl)和3-岩藻糖基乳糖(3
’‑
fl)
136.质粒prs305-p
gal1-futbc-t
cyc1
是将启动子p
gal1
、α-1,2-岩藻糖基转移酶基因futbc和终止子t
cyc1
插入载体质粒prs305中构建得到,构建方法可参考文献:xu,m.,et al.,improved production of 2
′‑
fucosyllactose in engineered saccharomyces cerevisiae expressing a putativeα-1,2-fucosyltransferase from bacillus cereus.microbial cell factories,2021.20(1):p.165。
137.质粒prs305-p
gal1-fut3bc-t
cyc1
是将启动子p
gal1
、α-1,3-岩藻糖基转移酶基因
fut3bc(seq id no.2)和终止子t
cyc1
插入载体质粒prs305中构建得到,构建方法与质粒prs305-p
gal1-futbc-t
cyc1
的构建方法相同,不同之处在于使用fut3bc基因替换了futbc基因。
138.1、以酿酒酵母w303-1a基因组dna为模板,扩增得到半乳糖代谢基因gal上下游同源臂以及启动子p
tdh3
和终止子t
cyc1
,以克鲁维酵母基因组dna为模板,扩增得到乳糖透性酶lac12编码基因,以质粒prs303为模板,扩增得到his3筛选marker基因。
139.pcr扩增引物序列如下:
140.up-gal-f:5
′‑
ggggaaacttaaagaaattc-3
′
;
141.up-gal-his-r:5
′‑
agtgtactagagtcaagagtcgtagtggag-3
′
;
142.his-f:5
′‑
acgactcttgactctagtacactctatatttttttatg-3
′
;
143.his-r:5
′‑
tattgtcagtctacataagaacacctttg-3
′
;
144.teft-his-f:5
′‑
gttcttatgtagactgacaataaaaagattcttg-3
′
;
145.teft-tdh3p-r:5
′‑
gataatgacagtatagcgaccagc-3
′
;
146.tdh-lac12-f:5
′‑
tcgctatactgtcattatcaatactgcc-3
′
;
147.cyc1t-r:5
′‑
aagtatacgcaaattaaagccttcg-3
′
;
148.down-gal-f:5
′‑
gctttaatttgcgtatacttcttttttttactttg-3
′
;
149.down-gal-r:5
′‑
gtttcaagacggcaatc-3
′
。
150.首先将his3、p
tdh3-lac12-t
cyc1
以1:1的摩尔比融合,所得到的pcr产物进一步与gal代谢基因上下游同源臂1:3:1摩尔比融合,得融合pcr产物gal1/7/10δ::p
tdh3-lac12,该产物以his3为筛选标记;将gal1/7/10δ::p
tdh3-lac12转化至工程菌kα-ege2中,通过组氨酸缺陷的sc固体培养基筛选,得工程菌kα-ege4-l,该菌株可吸收胞外的乳糖。
151.将质粒prs305-p
gal1-futbc-t
cyc1
和质粒prs305-p
gal1-fut3bc-t
cyc1
分别转化至工程菌kα-ege4-l中,通过在缺少亮氨酸的sc固体培养基上进行筛选,分别得到菌株kα-ege4-2fl和kα-ege4-3fl。
152.2、以半乳糖诱导型启动子p
gal
表达α-1,2-岩藻糖基转移酶基因futbc,2
’‑
fl合成途径的菌株fl06作为kα-ege4-2fl菌株的对照,该菌株的构建方法可参考文献:xu,m.,et al.,improved production of 2
′‑
fucosyllactose in engineered saccharomyces cerevisiae expressing a putativeα-1,2-fucosyltransferase from bacillus cereus.microbial cell factories,2021.20(1):p.165。
153.以半乳糖诱导型启动子p
gal
表达3
’‑
fl合成途径的菌株fl303作为kα-ege4-3fl菌株的对照,该菌株的构建方法与菌株fl06的构建方法相同,不同之处仅在于用α-1,3-岩藻糖基转移酶基因fut3bc(seq id no.2)替换了α-1,2-岩藻糖基转移酶基因futbc。
154.3、发酵生产2
’‑
fl的过程如下:
155.将菌株kα-ege4-2fl和对照菌株fl06的菌液接种至带有5ml的ypd液体培养基(添加20g/l葡萄糖和0.06g/l adenine sulfate)中,在30℃,200rpm下震荡培养24h后,接种于带有20mlypd液体培养基(添加20g/l葡萄糖和0.06g/l adenine sulfate)中,调整接种量,使发酵液的初始od为约0.2,继续在摇床中30℃,200rpm震荡培养24h后,菌株kα-ege4-2fl补加30g/l的葡萄糖,4g/l的乳糖,其对照菌株fl06补加30g/l的半乳糖,4g/l的乳糖,发酵产物中的2
’‑
fl产量如图6所示。
156.由图6可知,菌株kα-ege4-2fl中2
’‑
fl产量在48h时即达到最高值3.37g/l,该产量为48h时对照菌株fl06产量的2.6倍,明显提高了2
’‑
fl合成效率。
157.4、发酵生产3
’‑
fl的过程如下:
158.将菌株kα-ege4-2fl和对照菌株fl303的菌液接种至带有5ml的ypd液体培养基(添加20g/l葡萄糖和0.06g/l adenine sulfate)中,在30℃,200rpm下震荡培养24h后,接种于带有20ml ypd液体培养基(添加20g/l葡萄糖和0.06g/l adenine sulfate)中,调整接种量,使发酵液的初始od为约0.2,继续在摇床中30℃,200rpm震荡培养24h后,菌株kα-ege4-3fl补加30g/l的葡萄糖,2g/l的乳糖,其对照菌株fl303补加30g/l的半乳糖,2g/l的乳糖,发酵产物中的3
’‑
fl产量如图7所示。
159.由图7可知,菌株kα-ege4-3fl在96h时合成的3
’‑
fl产量为2.36g/l,该产量96h对照菌株fl303产量的2.5倍,明显提高了3
’‑
fl合成效率。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1