高纯度啶酰菌胺晶型I的制备方法与流程
高纯度啶酰菌胺晶型i的制备方法
技术领域
1.本发明涉及一种高纯度啶酰菌胺晶型i的制备方法。
背景技术:
2.啶酰菌胺的杀菌谱较广,几乎对所有类型的真菌都有活性,可用于防止白粉病、灰霉病、根腐病等,果树、葡萄、蔬菜和大田作物等均可使用。不同晶型啶酰菌胺其在制剂领域的应用范围不同,啶酰菌胺晶型i可用于制备水中可分散的喷雾干燥式或挤出式颗粒剂。
3.中国专利cn109071445a中公开了一种啶酰菌胺晶型i的制备方法,该方法包括先将啶酰菌胺无水物的多晶型ii溶解溶于第一溶剂中,将所得溶液与水合并,从溶剂混合物中分离固体,干燥后得到啶酰菌胺无水物的多晶型i。
4.中国专利cn114181141a中公开了另一种啶酰菌胺晶型i的制备方法,该方法是将无水啶酰菌胺溶于非水溶性有机溶剂中得到溶液,再将该溶液加入热水中闪蒸溶剂,得到悬浮于水中的啶酰菌胺晶型i湿品,经冷却、过滤、干燥后得到啶酰菌胺晶型i产品的方法。该方法需在100℃以上高温条件下反应,不仅能耗高,且存在安全风险;同时高温过程产业化相对复杂,难以形成规模化生产。
5.现有啶酰菌胺晶型i的制备方法存在晶型不稳定、工艺操作难度大等问题。
技术实现要素:
6.鉴于以上问题,本发明旨在提供一种新的制备啶酰菌胺晶型i的方法,通过该方法能够获得高晶型纯度和稳定性的啶酰菌胺晶型i。
7.具体地,本发明提供一种制备啶酰菌胺晶型i的方法,该方法包括以下步骤:
8.a.将啶酰菌胺固体加入到有机溶剂中,在控温条件下搅拌至完全溶解;
9.b.加入纯化水,在控温条件下继续搅拌溶清;
10.c.过滤;
11.d.进一步加入纯化水,进一步回流溶清;
12.e.降温养晶;
13.f.过滤,洗涤,干燥。
14.在一些具体实施方式中,的有机溶剂为丙酮或四氢呋喃。
15.在一些具体实施方式中,步骤a中所用有机溶剂与啶酰菌胺固体的质量比为(10~15):1。
16.在一些具体实施方式中,步骤a中的控温温度为28~32℃。
17.本技术发明人在生产过程中发现,如果仅使用有机溶剂进行结晶,在结晶过程中,因丙酮或四氢呋喃等有机溶剂的挥发会导致啶酰菌胺晶型ii的析出,进而导致啶酰菌胺晶型i的收率和晶型纯度降低,所获晶型i的纯度低于95%。另外,晶型ii的混杂会导致晶型i在存储中更容易发生转晶,晶型稳定性降低。
18.为了解决上述有机溶剂挥发而导致晶型ii析出的问题,本技术发明人在步骤a后
尝试加水。进一步地,本技术发明人发现,在步骤a之后,如果一次加水过多会导致啶酰菌胺的溶解度降低,析出晶体,不能充分溶清,进而导致杂质去除不充分,降低目标晶型的质量。另一方面,如果此阶段通过升温来促进溶清,则溶清之后的过滤是热过滤,操作不便。
19.因此,本发明在步骤a之后设置步骤b:加入纯化水,并在控温条件下继续搅拌溶清。在步骤b中,为了防止啶酰菌胺因溶解度降低而析出晶体,需要控制纯化水的加入量。
20.在一些具体实施方式中,步骤b中所用纯化水与步骤a中的啶酰菌胺固体的质量比为(1~2):1。
21.在一些具体实施方式中,步骤b中的控温温度为28~32℃。
22.步骤b之后,为了除去不溶性杂质,进行步骤c:过滤。优选地,过滤所用的滤膜或滤芯孔径为0.22μm以下。
23.为了获得高纯度的晶型i,本发明的方法在过滤后进一步往滤液中加入纯化水,并在回流条件下溶清。本技术发明人发现,回流溶清可以防止晶ii的形成,使后续降温析晶过程全部得到晶型i的产品。
24.在一些具体实施方式中,步骤c中所用纯化水与步骤a中的啶酰菌胺固体的质量比为(5~10):1。
25.在一些具体实施方式中,步骤c中的回流温度为60~62℃。
26.在一些具体实施方式中,步骤c中的回流溶清时间为6~8小时,时间太短的话,晶型纯度不够。
27.在一些具体实施方式中,步骤e中降温至15~35℃,养晶时间为0.5~2小时。
28.优选地,步骤e的降温养晶分两段进行,先降温至一定温度养晶,让结晶长大,然后进一步降温至更低温度养晶,以析出更多的结晶,提高收率。这样的分段降温养晶过程析出的晶体粒度更大,有利于过滤、洗涤和干燥过程。
29.更优选地,步骤e中先降温至30~35℃养晶0.5~1小时,然后降温至15~20℃养晶0.5~1小时。
30.在优选的实施方式中,步骤f中的干燥分段进行,先低温干燥去除有机溶剂,然后在更高温度下干燥除水。由此可实现有机溶剂和水的分阶段去除,从而避免干燥过程中的转晶现象,有效控制晶型稳定性。
31.在更优选的实施方式中,步骤f中的干燥分两段进行,先35~40℃干燥,再升温至65~70℃干燥,。
32.本发明方法反应条件温和、操作简单、节能环保,制备出的啶酰菌胺晶型i纯度和稳定性高、含量高。
附图说明
33.图1为本技术实施例1制备的啶酰菌胺晶型i的dsc曲线。
34.图2为本技术实施例1制备的啶酰菌胺晶型i的x射线粉末衍射谱图。
具体实施方式
35.下面结合具体实施例进一步对本发明进行详细说明,但本发明不限于这些具体实施例,任何在本发明主旨范围内的修改或变更都落入本技术范围。
36.实施例1
37.取16g啶酰菌胺晶型ii原料,加入160g丙酮,控温32℃,搅拌0.5小时至完全溶解,加入16g纯化水,继续搅拌溶解0.5小时。使用0.22μm除菌滤膜过滤,用40g丙酮洗涤。
38.合并滤液及洗液,控温60℃回流条件下加入80g纯化水,继续保持60℃回流并搅拌溶解8小时。
39.缓慢降温至35℃,养晶0.5小时;继续降温至15℃,养晶0.5小时;过滤,用32g纯化水洗涤,抽干;35℃鼓风干燥1小时,65℃鼓风干燥1小时,得白色固体啶酰菌胺晶型i产品。
40.通过卡尔费休(kf)滴定法检测产品的水分、通过高效液相色谱法检测产品含量,结果如表1所示。
41.高效液相色谱条件:
42.色谱柱:c18柱250mm
×
4.6mm
×
5um;流动相:乙腈:水(甲酸调ph约为3.7-4.1)=80:20;柱温:30℃;流速:1.0ml/min;波长:254nm。
43.通过差示扫描量热仪(dsc)检测产品的晶型纯度,测定条件如下,结果如图1和表1所示。
44.升温区间:20-120℃;升温速度为4℃/min;
45.升温区间:120-170℃;升温速度为1℃/min。
46.测定产品的x射线粉末衍射图谱,测定条件如下,结果如图2所示。
47.扫描2θ:5.0~100.0
°
,步进:0.02
°
/sec,cu(45kv,200ma)。
48.实施例2
49.取16g啶酰菌胺混合晶型原料,加入240g丙酮,控温28℃,搅拌0.5小时至完全溶解,加入32g纯化水,继续搅拌溶解0.5小时。使用0.22μm除菌滤膜过滤,用40g丙酮洗涤。
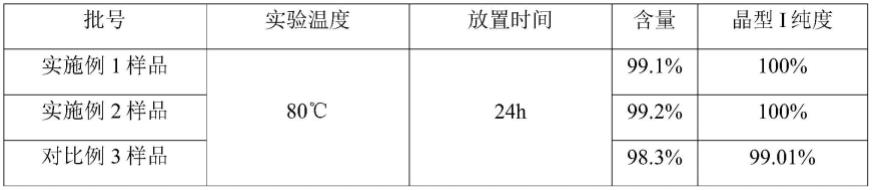
50.合并滤液及洗液,控温62℃回流条件下加入160g纯化水,继续保持62℃回流并搅拌6小时。
51.缓慢降温至30℃,养晶1小时;继续降温至20℃,养晶1小时;过滤,用32g纯化水洗涤,抽干;40℃鼓风干燥1小时,70℃鼓风干燥1小时,得白色固体啶酰菌胺晶型i产品。与实施例1同法检测产品的水分、晶型纯度、含量质量指标,结果如表1所示。
52.对比例1
53.取16g啶酰菌胺晶型ii原料,加入160g丙酮,控温32℃,搅拌0.5小时至完全溶解,在未加入水条件下,继续搅拌溶解0.5小时。使用0.22μm除菌滤膜过滤,用40g丙酮洗涤。
54.在膜过滤过程中,由于丙酮挥发造成部分晶体析出,将此晶体过滤收集,按照实施例1的方法干燥并测定产品的dsc曲线,晶型i纯度为91.61%。
55.对比例2
56.取16g啶酰菌胺晶型ii原料,加入160g丙酮,控温32℃,搅拌0.5小时至完全溶解,在加入水96g条件下,继续搅拌溶解0.5小时。使用0.22μm除菌滤膜过滤,过滤过程晶体析出,堵塞过滤器,影响过滤过程。
57.对比例3
58.取16g啶酰菌胺晶型ii原料,加入160g丙酮,控温32℃,搅拌0.5小时至完全溶解,加入16g纯化水,继续搅拌溶解0.5小时。使用0.22μm除菌滤膜过滤,用40g丙酮洗涤。
59.合并滤液及洗液,控温60℃回流条件下加入80g纯化水,继续保持60℃回流4小时。
60.缓慢降温至35℃,养晶0.5小时;继续降温至15℃,养晶0.5小时;过滤,用32g纯化水洗涤,抽干;35℃鼓风干燥1小时,65℃鼓风干燥1小时,得白色固体啶酰菌胺晶型i产品。与实施例1同法检测产品的水分、晶型纯度、含量质量指标,结果如表1所示。
61.表1
62.批号水分含量晶型i纯度实施例10.15%99.3%100%实施例20.14%99.2%100%对比例30.25%98.5%99.55%
63.由表1的结果可知,实施例1和2产品含量和晶型i纯度更高,产品晶型i纯度达到100%,不存在混合晶型。
64.实施例3晶型热稳定性考察
65.分别称取5g实施例1、实施例2及对比例3的产品样品,于80℃鼓风烘箱中干燥24h。分别取样,检测晶型i纯度,结果如表2所示。
66.表2
[0067][0068]
由表2的结果可知,通过本发明方法获得的100%晶型i纯度的产品在高温干燥后晶型i纯度仍然保持在100%,具有良好的稳定性。晶型i纯度未达到100%的对比例3的样品在高温干燥后晶型i纯度会降低。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1