胆酸的合成方法与流程
1.本发明属于制药技术领域,具体涉及一种胆酸的合成方法。
背景技术:
2.胆酸,化学名为cholic acid,化学式为c
24h40
o5,其结构式如下:
[0003][0004]
胆汁酸是胆汁中存在的一类胆烷酸的总称,人类胆汁中存在的胆汁酸主要有胆酸 (ca)、鹅脱氧胆酸(cdca)、脱氧胆酸(dca)和石胆酸(lca)。
[0005]
胆酸可以用于生化研究,是一种医药中间体。cn201710404532.5公开了一种以胆酸为原料合成石胆酸的方法、cn2017102663050.0公开了一种以胆酸为原料合成熊去氧胆酸的方法、cn2016107273812公开了一种以胆酸为原料制备甘氨胆酸多克隆抗体的方法 cn2021102688904公开了一种以胆酸为起始原料合成脱氧胆酸的方法,熊去氧胆酸、石胆酸等都是可以作为治疗药物使用,从上可以看出胆酸是重要的医药中间体。
[0006]
另外,胆酸钠是利胆药,能够用于治疗胆囊炎、胆汁缺乏、肠道消化不良等症。
[0007]
目前市场上胆酸均为动物牛或羊的内脏提取精制而成,不可避免地存在病毒传染风险。
技术实现要素:
[0008]
本发明的目的在于提供一种反应条件温和的化学合成方法生产胆酸,以解决通过动物内脏提取胆酸带来的病毒传染的风险。
[0009]
为了实现上述目的,本发明提供一种胆酸的合成方法,所述胆酸的结构式为式ca所示,所述合成方法包括以下步骤:
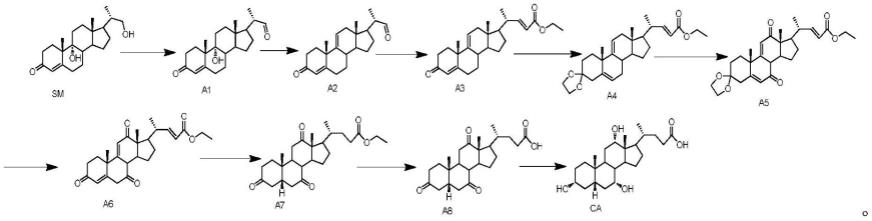
[0010]
(1)将式sm所示化合物进行第一氧化反应得到式a1所示化合物;
[0011]
(2)将所述式a1所示化合物进行消除反应得到式a2所示化合物;
[0012]
(3)将所述式a2所示化合物进行wittig或者wittig-horner反应得到式a3所示化合物;
[0013]
(4)将所述式a3所示化合物进行缩酮保护反应得到式a4所示化合物;
[0014]
(5)将所述式a4所示化合物进行第二氧化反应得到式a5所示化合物;
[0015]
(6)将所述式a5所示化合物进行第一水解反应得到式a6所示化合物;
[0016]
(7)将所述式a6所示化合物进行氢化还原反应得到式a7所示化合物;
[0017]
(8)将所述式a7所示化合物进行第二水解反应得到式a8所示化合物;
[0018]
(9)将所述式a8所示化合物进行选择性还原反应得到胆酸;
[0019]
上述九步反应在的完整反应式如下:
[0020][0021]
在一种具体的实施方式中,步骤(1)中,所述第一氧化反应是在溶剂二氯甲烷中,使所述式sm所示化合物与氧化剂tempo和8%次氯酸钠在室温下进行反应,或者,所述第一氧化反应在溶剂丙酮中,使所述式sm所示化合物与氧化剂琼斯试剂在0~10℃下进行反应。
[0022]
在一种具体的实施方式中,步骤(2)中,所述消除反应是在溶剂水中,使所述a1 所示化合物与浓硫酸和冰醋酸在20~50℃下进行反应;或者所述消除反应是在溶剂二氯甲烷或者三氯甲烷中,使所述式a1所示化合物与醋酸酐和浓硫酸在30~40℃下反应。
[0023]
在一种具体的实施方式中,步骤(3)中,所述wittig反应是在溶剂甲苯或四氢呋喃中,使式a2所示化合物和wittig试剂乙氧甲酰基亚甲基三苯基膦混合得到反应物体系,使所述反应物体系升温至回流状态进行反应;或者所述wittig-horner反应是在溶剂四氢呋喃中,使式a2所示化合物与碱和膦酰基乙酸三乙酯在20~66℃下进行反应,其中,所述碱为氢化钠、叔丁醇钾、叔丁醇钠、甲醇钠、甲醇钾、乙醇钠、乙醇钾中的一种。
[0024]
在一种具体的实施方式中,步骤(4)中,所述缩酮保护反应是在溶剂甲苯中,使式 a3所示化合物与缩酮反应试剂乙二醇和催化剂对甲苯磺酸混合后,使体系升温至回流状态进行反应;或者所述缩酮保护反应是使a3所示化合物与缩酮反应试剂乙二醇、缩酮脱水剂原甲酸三乙酯和催化剂对甲苯磺酸在35~80℃下进行反应。
[0025]
在一种具体的实施方式中,步骤(5)中,所述第二氧化反应是在溶剂以丙酮或者乙腈中,使式a4所示化合物与氧化剂氧化铬和n-羟基邻苯二甲酰亚胺的混合物或者氧化剂铬酸吡啶翁和n-羟基邻苯二甲酰亚胺的混合物在20~35℃下进行氧化反应。
[0026]
在一种具体的实施方式中,步骤(6)中,所述第一水解反应是在溶剂甲醇中,使式 a5所示化合物在酸性条件下在20~65℃下进行反应,其中,所述第一水解反应采用的酸为盐酸或者硫酸。
[0027]
在一种具体的实施方式中,步骤(7)中,所述氢化还原反应是在溶剂无水乙醇和吡啶的混合物中,以钯质量含量为8~12%的钯碳为催化剂,使式a6所示化合物与氢气在预设条件下进行反应,其中,所述预设条件包括:氢化还原反应压力为0.4~0.45mpa,反应温度为30~35℃。
[0028]
在一种具体的实施方式中,步骤(8)中,所述第二水解反应是在特定溶剂中,使式 a7所示化合物的侧链在碱性条件下在40~60℃下进行反应,其中,所述特定溶剂为甲醇、乙醇、丙酮、四氢呋喃中的一种或者二氯甲烷和甲醇的混合物或者二氯甲烷和乙醇的混合物,所述第二水解反应采用的碱为氢氧化钠或者氢氧化钾。
[0029]
在一种具体的实施方式中,步骤(9)中所述选择性还原反应是在溶剂四氢呋喃中,使式a8所示化合物与还原剂三叔丁氧基氢化铝锂在20~25℃下进行反应。
[0030]
本发明的有益效果至少包括:
[0031]
本发明使式sm所示化合物依次通过第一氧化反应、消除反应、wittig反应、缩酮保
护反应、第二氧化反应、第一水解反应、氢化还原反应、第二水解反应、选择性还原反应得到式ca所示化合物胆酸,提供了一种以植物源发酵产物式sm所示化合物为原料合成胆酸的化学合成方法,且具有反应条件温和的优点,能够克服现有技术通过动物内脏提取胆酸带来的病毒传染的风险。
具体实施方式
[0032]
一种胆酸的合成方法,所述胆酸的结构式为式ca所示,所述合成方法包括以下步骤:
[0033]
(1)将式sm所示化合物进行第一氧化反应得到式a1所示化合物;
[0034]
(2)将所述式a1所示化合物进行消除反应得到式a2所示化合物;
[0035]
(3)将所述式a2所示化合物进行wittig反应或者wittig-horner反应得到式a3所示化合物;
[0036]
(4)将所述式a3所示化合物进行缩酮保护反应得到式a4所示化合物;
[0037]
(5)将所述式a4所示化合物进行第二氧化反应得到式a5所示化合物;
[0038]
(6)将所述式a5所示化合物进行第一水解反应得到式a6所示化合物;
[0039]
(7)将所述式a6所示化合物进行氢化还原反应得到式a7所示化合物;
[0040]
(8)将所述式a7所示化合物进行第二水解反应得到式a8所示化合物;
[0041]
(9)将所述式a8所示化合物进行选择性还原反应得到胆酸;
[0042]
上述九步反应在的完整反应式如下:
[0043][0044]
在本实施例中,所述式sm所示化合物为植物源发酵产物,可以自己制备也可以通过商业采购。自己制备合成路线为:
[0045][0046]
在一种可选方式中,步骤(1)中所述第一氧化反应采用的氧化剂为tempo和8%次氯酸钠,所述式sm所示化合物与氧化剂tempo和氧化剂8%次氯酸钠的质量之比为 1:(0.1~0.13):(2~2.2)。
[0047]
在本实施例中,8%次氯酸钠是指有效氯为8%的次氯酸钠。
[0048]
优选地,步骤(1)中所使用的溶剂为二氯甲烷,所述式sm所示化合物与溶剂二氯甲烷的质量体积比为1g:(7.5~8.5)ml。
[0049]
优选地,步骤(1)中所述第一氧化反应的温度为室温,所述第一氧化反应的时间为 2.5~4h。
[0050]
所述第一氧化反应具体为:以二氯甲烷为溶剂,搅拌下加入9α-羟基ba(式sm所示)和氧化剂tempo,然后再滴加氧化剂8%次氯酸钠,滴加完成后反应2.5-4h,tlc 检测反应至完全,加入5%亚硫酸钠溶液,用淀粉碘化钾试纸测无氧化性为止。体系搅拌 20分钟左右,静置30分钟左右,分液,收集有机相,有机相中加入水,搅拌20分钟左右,静置30分钟左右,分液,收集有机相;40℃热水浴下减压浓缩蒸发二氯甲烷,甲醇置换,浓缩至粘稠状态为止,再降温、过滤、干燥得到式a1所示化合物。
[0051]
在另一种可选方式中,步骤(1)中所述第一氧化反应采用的溶剂为丙酮,所述氧化剂为琼斯试剂,所述第一氧化反应温度为0~10℃,所述第一氧化反应时间为4~8h。
[0052]
优选地,所述式sm所示化合物与所述氧化剂琼斯试剂的质量体积比为1g: (1~1.2)ml,较佳为1:1。
[0053]
优选地,所述式sm所示化合物与所述溶剂丙酮的质量体积比为1g:(9~11)ml,较佳为1:10。
[0054]
所述第一氧化反应具体为:以丙酮为溶剂,搅拌下加入9α-羟基ba(式sm所示),将体系降温至0-10℃,控温滴加琼斯试剂,保温搅拌4~8h,tlc检测反应至完全。加入异丙醇淬灭反应,将体系缓慢加入到冰水中水析,降温至0-10℃,过滤,干燥得到式a1所示化合物。
[0055]
在一种可选方式中,步骤(2)中所述消除反应在酸性条件下进行,所述消除反应中采用的酸为浓硫酸和冰醋酸,所述消除反应的温度为20~50℃,所述消除反应的时间为 2.5-4h。
[0056]
优选地,所述消除反应的温度为35~40℃。
[0057]
优选地,步骤(2)中,所述式a1所示化合物与浓硫酸的质量比为1:(3.5~3.8),所述式a1所示化合物与冰醋酸的质量体积比为1g:(1~1.2)ml。
[0058]
所述消除反应具体为:在反应器中加入水,冰水浴降温至0~10℃,搅拌下控温t≤ 40℃,滴加浓硫酸,然后加入冰醋酸和式a1所示化合物,将体系控温至20~50℃反应 2.5~4h,tlc检测反应至完全。将体系加入到冰水浴中,加完后搅拌30分钟以上,过滤,用水淋洗至中性,将滤饼用二氯甲烷溶解,分液,收集有机相,减压浓缩,甲醇置换,最终浓缩至粘稠状态,过滤,滤饼干燥至合格,得到式a2所示化合物。
[0059]
在另一种可选方式中,所述消除反应以二氯甲烷或者三氯甲烷为溶剂,使式a1所示化合物与醋酸酐和浓硫酸在30~40℃反应3~5h。
[0060]
优选地,所述式a1所示化合物与醋酸酐的质量体积比为1g:(1~1.2)ml,所述醋酸酐与浓硫酸的体积比为1:(0.38~0.4)。
[0061]
优选地,所述式a1所示化合物与溶剂二氯甲烷或者三氯甲烷的质量体积比为1g: (5.8~6.2)ml。
[0062]
所述消除反应具体为:在搅拌状态下,将式a1所示化合物加入到溶剂二氯甲烷或者三氯甲烷中,降温至0-10℃,滴加醋酸酐,加完后搅拌10~20分钟,再滴加浓硫酸,滴加完成后升温至30~40℃,保温搅拌3-5h,tlc检测反应至完全。在体系中加水淬灭,分液,收集有机相,减压浓缩,甲醇置换,最终浓缩至粘稠状态,过滤,滤饼干燥至合格,得到式a2所示化合物。
[0063]
在一种可选方式中,式a2所示化合物进行wittig反应得到式a3所示化合物,进行 wittig反应,以甲苯或者四氢呋喃为溶剂,以乙氧甲酰基亚甲基三苯基膦为wittig试剂,使
式a2所示化合物和wittig试剂乙氧甲酰基亚甲基三苯基膦升温至回流状态进行反应,所述wittig反应的时间为10~16h。
[0064]
优选地,所述式a2所示化合物和wittig试剂乙氧甲酰基亚甲基三苯基膦的质量比为 1:(1.8~2.4),较佳地,所述式a2所示化合物和wittig试剂乙氧甲酰基亚甲基三苯基膦的质量比为1:2。
[0065]
所述wittig反应具体为:在搅拌状态下,将式a2所示化合物和乙氧甲酰基亚甲基三苯基膦加入到溶剂甲苯或者四氢呋喃中,将体系升温至回流状态反应10~16h,tlc检测反应至完全,降至室温。在体系中加入氯化锌溶液,于25~30℃下保温12h进行沉淀反应,抽滤,将沉淀物过滤,滤液进行分液,再次用氯化锌溶液重复操作一次。过滤后分液,收集有机相,减压浓干,过滤,干燥得到式a3所示化合物。
[0066]
优选地,所述沉淀反应加入的金属离子络合剂为氯化锌溶液,所述氯化锌溶液的质量分数为35%~45%,较佳地,所述氯化锌溶液的质量分数为36%~40%。
[0067]
优选地,所述沉淀反应的温度为25~30℃,反应时间为10~16h。
[0068]
以用氯化锌溶液为金属离子络合剂通过沉淀反应以除去副产物三苯基氧膦,得到较纯的目标产物。
[0069]
在另一种可选方式中,式a2所示化合物进行wittig-horner反应得到式a3所示化合物,步骤(3)wittig-horner反应以四氢呋喃为溶剂,以碱和膦酰基乙酸三乙酯的混合物为wittig-horner试剂,在20~66℃下反应1~3h直至反应完全;其中,所述碱为氢化钠、叔丁醇钾、叔丁醇钠、甲醇钠、甲醇钾、乙醇钠、乙醇钾中的一种。
[0070]
优选地,所述式a2所示化合物与碱的摩尔比为:1:(1.5~2.0),所述式a2所示化合物与膦酰基乙酸三乙酯的质量体积比为1g:(0.9~1.1)ml。
[0071]
所述wittig-horner反应具体为:搅拌状态下,将碱氢化钠/叔丁醇钾/叔丁醇钠/甲醇钠/甲醇钾/乙醇钠/乙醇钾中的一种加入到溶剂四氢呋喃中,有气泡冒出后,使用氮气保护,室温下滴加膦酰基乙酸三乙酯,搅拌20~30分钟,滴加式a2所示化合物和四氢呋喃的混合液,滴加完成后控温20~66℃反应1~3h,tcl跟踪直至反应完全,得到反应产物,经ph值调节、减压浓干四氢呋喃、水析、降温、过滤、干燥得到式a3所示化合物。
[0072]
优选地,所述混合液中式a2所示化合物与四氢呋喃的质量体积比为1g:(9~12) ml。
[0073]
在一种可选方式中,步骤(4)中所述缩酮保护反应以甲苯为溶剂,以乙二醇为缩酮反应试剂,以对甲苯磺酸为催化剂,将式a3所示化合物与甲苯、乙二醇和对甲苯磺酸混合后,使体系升温至回流状态进行反应,所述缩酮保护反应时间为20~28h。
[0074]
优选地,所述缩酮保护反应中的式a3所示化合物与催化剂对甲苯磺酸的质量比为 1:(0.098~0.018),更为较佳的为0.015。
[0075]
优选地,所述所述缩酮保护反应中的式a3所示化合物与所述溶剂甲苯的质量体积为 1g:(9~11)ml,较佳地为1:10g/ml;所述所述缩酮保护反应中的式a3所示化合物与所述反应试剂乙二醇的质量体积为1g:(3~4)ml,较佳地为1:2g/ml。
[0076]
所述缩酮保护反应具体为:将甲苯、乙二醇、甲苯磺酸(pts)和式a3所示化合物混合后,将体系升温至回流状态反应20~28h,tlc检测反应至完全。降温到室温,加入少量三乙胺调节ph=7-8,水洗,收集有机相,将甲苯层减压浓缩至粘稠状态,加入石油醚进行置
换,降温至0~10℃,过滤,干燥得到式a4所示化合物。
[0077]
在另一种可选方式中,步骤(4)中所述缩酮保护反应以乙二醇为缩酮反应试剂,以原甲酸三乙酯为缩酮脱水剂,以对甲苯磺酸为催化剂,所述缩酮保护反应的温度为 35~80℃,反应时间为5~8h。
[0078]
优选地,所述缩酮保护反应的温度为40~45℃。
[0079]
优选地,所述缩酮保护反应中的式a3所示化合物与催化剂对甲苯磺酸的质量比为 1:(0.098~0.018),更为较佳的为0.015。
[0080]
所述缩酮保护反应具体为:将缩酮反应试剂乙二醇、缩酮脱水剂原甲酸三乙酯和催化剂对甲苯磺酸混合后,搅拌状态下加入式a3所示化合物,将体系升温至35~80℃,反应5~8h,tcl检测反应至完全,降温至室温,加入三乙胺调节ph值至7~8,然后经水析、继续降温至0~10℃、过滤得到滤饼,所述滤饼用甲醇重结晶后,降温至室温,过滤、干燥得到式a4所示化合物。
[0081]
优选地,步骤(5)中所述第二氧化反应以丙酮或者乙腈为溶剂,以氧化铬和n-羟基邻苯二甲酰亚胺的混合物或者氯铬酸吡啶翁和n-羟基邻苯二甲酰亚胺为氧化剂,使式a4 所示化合物与氧化剂在20~35℃下反应20~25h。
[0082]
优选地,所述第二氧化反应的温度为20~25℃。
[0083]
当温度高于氧化反应的温度时,会导致3位水解,无法得到目标产物式a5所示化合物。
[0084]
优选地,所述氧化剂为氧化铬和n-羟基邻苯二甲酰亚胺的混合物,所述式a4所示化合物、氧化铬和n-羟基邻苯二甲酰亚胺加入的重量比为1:(1.9~2.2): (0.7~0.76);更为优选地,所述式a4所示化合物、氧化铬和n-羟基邻苯二甲酰亚胺(nhpi)加入的重量比为1:2:0.72。
[0085]
优选地,所述氧化剂为氯铬酸吡啶翁和n-羟基邻苯二甲酰亚胺的混合物,所述式a4 所示化合物、氧化铬和n-羟基邻苯二甲酰亚胺加入的重量比为1:(2.3~2.6): (0.7~0.76);更为优选地,所述式a4所示化合物、氯铬酸吡啶翁和n-羟基邻苯二甲酰亚胺加入的重量比为1:2:0.72。
[0086]
优选地,所述第二氧化反应的溶剂为丙酮时,所述式a4所示化合物与所述丙酮的质量体积比为1g:(13~16)ml。
[0087]
优选地,所述第二氧化反应的溶剂为乙腈时,所述式a4所示化合物与所述乙腈的质量体积比为1g:(18~21)ml。
[0088]
所述第二氧化反应具体为:将丙酮/乙腈和水加入反应器,搅拌下加入式a4所示化合物、一半氧化铬或者氯铬酸吡啶翁以及n-羟基邻苯二甲酰亚胺(nhpi),控温 20~35℃,保温搅拌3h后,再加入另一半氧化铬或者氯铬酸吡啶翁,保温搅拌20~25h, tcl检测反应至完全,然后加入异丙醇淬灭反应,将丙酮浓干,加入二氧甲烷提取铬酸,分液,有机相用水水洗一次,然后收集有机相,将所述有机相减压浓缩,甲醇置换,降温,过滤得到式a5所示化合物。
[0089]
优选地,所述第一水解反应包括:以甲醇为溶剂,使式a5所示化合物在酸性条件下发生第一水解反应生成式a6所示化合物,其中,所述第一水解反应采用的酸为盐酸或者硫酸,所述第一水解反应温度为20~65℃,反应时间为3~5h。
[0090]
优选地,所述第一水解反应温度为50~55℃。
[0091]
优选地,所述式a5所示化合物与盐酸的质量体积比为1g:(0.45~0.6)ml。
[0092]
所述第一水解反应具体为:将甲醇、水和酸加入反应器,搅拌下加入式a5所示化合物,将体系升温至20~65℃,反应3~5h,tcl检测至反应完全,将体系降温至30℃以下,用饱和碳酸氢钠溶液调节ph至6~7,过滤,干燥得到式a6所示化合物。
[0093]
优选地,所述氢化还原反应包括:以无水乙醇和吡啶的混合物为溶剂,以钯质量含量为8~12%的钯碳为催化剂,使式a6所示化合物与氢气发生氢化还原反应生成式a7所示化合物,其中,所述氢化还原反应压力为0.4~0.45mpa,反应温度为30~35℃,反应时间为8~13h。
[0094]
优选地,所述无水乙醇和所述吡啶的体积比为1:(0.9-1.1)。
[0095]
优选地,所述氢化还原反应的溶剂还包括甲基咪唑,所述无水乙醇与所述吡啶和甲基咪唑的混合物的体积之比为1:(0.9-1.1),或者所述氢化还原反应的溶剂还包括 dmap,所述无水乙醇和吡啶的体积比为1:(0.9-1.1),所述dmap与所述无水乙醇的质量比积比为1g:(48-53)ml,所述式a6所示化合物与溶剂的质量体积比为1g:(9.5
‑ꢀ
10.5)ml。
[0096]
优选地,所述式a6所示化合物与催化剂钯碳的重量比为1:(0.09~0.12)。
[0097]
所述氢化还原反应具体为:将氢化还原反应的溶剂混合后,搅拌下加入式a6所示化合物和催化剂钯碳,在30~35℃用氢气加压至0.4~0.45mpa反应8~13h,tlc检测反应至完全;然后过滤去除钯碳,再减压浓缩溶剂,用水置换,水析,过滤,少量水淋洗,滤饼干燥至合格,得到式a7所示化合物。
[0098]
优选地,所述第二水解反应包括:在特定的溶剂中,使式a7所示化合物的侧链在碱性条件下发生第二水解反应生成式a8所示化合物,其中,所述第二水解反应采用的碱为氢氧化钠或者氢氧化钾,所述第二水解反应温度为40~60℃,反应时间为3~5h。
[0099]
所述特定的溶剂为甲醇、乙醇、丙酮、四氢呋喃中的一种或者二氯甲烷和甲醇的混合物或者二氯甲烷和乙醇的混合物。优选地,所述第二水解反应温度为50~55℃,反应时间为3h。
[0100]
优选地,所述式a7所示化合物与溶剂的质量体积比为1g:(5~10)ml。
[0101]
优选地,所述式a7所示化合物与氢氧化钠的质量比为1:(0.19~0.22),或者所述式a7所示化合物与氢氧化钾的质量比为1:(0.26~0.30)。
[0102]
需要说明的是所述侧链水解反应采用的碱也可以为氢氧化钠和氢氧化钾的混合物;需要说明的是,若加入的为固体碱,则还需要加入水溶解碱,当然也可以直接加入氢氧化钠溶液和氢氢化钾溶液。
[0103]
所述第二水解反应具体为:将特定的溶剂、水和碱氢氧化钠或者氢氧化钾混合后,搅拌状态下加入式a7所示化合物,然后将反应体系升温至50~55℃反应3~5h,tcl检测反应至完全,降温至30℃以下,用6mol/l盐酸调节ph值至2~3,减压浓干甲醇,水析,过滤,少量水淋洗,干燥得到式a8所示化合物。
[0104]
优选地,所述选择性还原反应包括:以四氢呋喃为溶剂,使式a8所示化合物与还原剂三叔丁氧基氢化铝锂发生选择性还原反应生成胆酸,其中,所述选择性还原反应的反应温度为20~25℃,反应时间为8~13h。
[0105]
优选地,所述式a8所示化合物与还原剂三叔丁氧基氢化铝锂的质量比为1: (2.8
~3.1),较佳地为1:3。
[0106]
优选地,所述式a8所示化合物与溶剂四氢呋喃的质量体积比为1g:(14~17)ml。
[0107]
所述选择性还原反应具体为:将式a8所示化合物与溶剂四氢呋喃混合后,氮气保护降温至0~5℃,分批缓慢加入还原剂三叔丁氧基氢化铝锂,加完后升温至20~25℃,反应 8~13h,tcl检测至反应完全,缓慢滴加1mol/l盐酸,调节ph值至2~3,减压浓干溶剂四氢呋喃,水析,过滤,少量水淋洗,滤饼用乙酸乙酯溶清后分液,重结晶精制,得到胆酸。
[0108]
需要说明的是,合成胆酸的原料式sm所示化合物9α-羟基ba为现有技术的产品,可以自制也可以商业采购。具体地,商业采购的供应商为深圳振强生物技术有限公司,自制的合成路线为:
[0109][0110]
本发明还提供一种胆酸中间体a8,所述胆酸中间体a8的结构式为式a8所示。
[0111]
本发明还提供一种胆酸中间体a8的制备方法,所述胆酸中间体a8的结构式为式a8 所示,所述制备方法包括以下步骤:在特定溶剂中,使式a7所示化合物在碱性条件发生侧链水解反应,反应完成后降至室温,经后处理,得到胆酸中间体a8,所述特定溶剂为甲醇、乙醇、丙酮、四氢呋喃中的一种或者二氯甲烷和甲醇的混合物或者二氯甲烷和乙醇的混合物。
[0112]
在该胆酸中间体a8的制备方法中,优选使式a6所示化合物在酸性条件下进行水解反应得到式a7所示化合物,更优选使式a5所示化合物在酸性条件下进行水解反应得到式a6所示化合物,更优选还包括使式a4所示化合物进行氧化反应得到式a5所示化合物,更优选还包括使式a3所示化合物进行缩酮保护反应得到式a4所示化合物,更优选还包括使式a2所示化合物进行wittig或者wittig-horner反应得到式a3所示化合物,更优选还包括使式a1所示化合物进行消除反应得到式a2所示化合物,更优选还包括使式 sm所示化合物进行氧化反应得到式a1所示化合物。
[0113]
本发明还提供一种胆酸中间体a8,包括采用如上所述胆酸中间体a8的制备方法制备得到。
[0114]
本发明还提供一种胆酸的合成方法,所述胆酸的分子式为式ca所示,所述合成路线包括采用如上所述的胆酸中间体a8制备所述胆酸;优选的,在溶剂存在下,使所述胆酸中间体a8与还原剂三叔丁氧基氢化铝锂进行选择性还原反应,得到所述胆酸。
[0115]
本发明还提供一种胆酸中间体a7,所述胆酸中间体a7的结构式为式a7所示。
[0116]
本发明还提供一种胆酸中间体a7的制备方法,所述胆酸中间体a7的结构式为式a7 所示,所述制备方法包括以下步骤:以无水乙醇和吡啶为溶剂,搅拌状态下加入式a6所示化合物和催化剂钯碳,在30~35℃用氢气加压至0.4~0.45mpa进行反应,反应完成后得到反应产物,所述反应产物经后处理后得到胆酸中间体a7。
[0117]
在该胆酸中间体a7的制备方法中,优选使式a5所示化合物在酸性条件下进行水解反应得到式a6所示化合物,更优选还包括使式a4所示化合物进行氧化反应得到式a5 所示化合物,更优选还包括使式a3所示化合物进行缩酮保护反应得到式a4所示化合物,更优选还包括使式a2所示化合物进行wittig或者wittig-horner反应得到式a3所示化合物,更优选还包括使式a1所示化合物进行消除反应得到式a2所示化合物,更优选还包括使式sm所示
化合物进行氧化反应得到式a1所示化合物。
[0118]
本发明还提供一种胆酸中间体a7,包括采用如上所述胆酸中间体a7的制备方法制备得到。
[0119]
本发明还提供一种胆酸的合成方法,所述胆酸的分子式为式ca所示,所述合成路线包括采用如上所述的胆酸中间体a7制备式a8所示化合物,再以所述式a8所示化合物制备所述胆酸;优选的,在溶剂中,使所述式a7所示化合物在碱存在的条件,进行水解反应,得到式a8所示化合物,再在溶剂存在下,使所述式a8所示化合物与还原剂三叔丁氧基氢化铝锂进行选择性还原反应,得到所述胆酸。
[0120]
本发明还提供一种胆酸中间体a6,所述胆酸中间体a6的结构式为式a6所示。
[0121]
本发明还提供一种胆酸中间体a6的制备方法,所述胆酸中间体a6的结构式为式a6 所示,所述制备方法包括以下步骤:以甲醇为溶剂,使式a5所示化合物在酸性条件下在 20~65℃下进行水解反应,反应完成后得到反应产物,所述反应产物经后处理后得到胆酸中间体a6。
[0122]
在该胆酸中间体a6的制备方法中,优选包括使式a4所示化合物进行氧化反应得到式a5所示化合物,更优选还包括使式a3所示化合物进行缩酮保护反应得到式a4所示化合物,更优选还包括使式a2所示化合物进行wittig或者wittig-horner反应得到式a3 所示化合物,更优选还包括使式a1所示化合物进行消除反应得到式a2所示化合物,更优选还包括使式sm所示化合物进行氧化反应得到式a1所示化合物。
[0123]
本发明还提供一种胆酸中间体a6,包括采用如上所述胆酸中间体a6的制备方法制备得到。
[0124]
本发明还提供一种胆酸的合成方法,所述胆酸的分子式为式ca所示,所述合成路线包括采用如上所述的胆酸中间体a6制备式a7所示化合物,再以所述式a7所示化合物制备式a8所示化合物,再以所述式a8所示化合物制备所述胆酸;优选的,在溶剂中,使所述a6所示化合物在催化剂钯碳的催化下进行氢化还原反应得到式a7所示化合物,再在溶剂中,使所述式a7所示化合物在碱存在的条件,进行水解反应,得到式a8所示化合物,再在溶剂存在下,使所述式a8所示化合物与还原剂三叔丁氧基氢化铝锂进行选择性还原反应,得到所述胆酸。
[0125]
本发明还提供一种胆酸中间体a5,所述胆酸中间体a5的结构式为式a5所示。
[0126]
本发明还提供一种胆酸中间体a5的制备方法,所述胆酸中间体a5的结构式为式a5 所示,所述制备方法包括以下步骤:在溶剂丙酮或者乙腈中,搅拌状态下加入式a4所示化合物、第一氧化剂和第二氧化剂,控制温度为20~25℃,保温搅拌第一预设时间后,再加入第一氧化剂,保温搅拌第二预设时间直至反应完全,然后加入异丙醇淬灭反应得到反应液,所述反应液经后处理得到胆酸中间体a5,其中,所述第一氧化剂为氧化铬或者氯铬酸吡啶翁,所述第二氧化剂为n-羟基邻苯二甲酰亚胺。
[0127]
在该胆酸中间体a5的制备方法中,优选包括使式a3所示化合物进行缩酮保护反应得到式a4所示化合物,更优选还包括使式a2所示化合物进行wittig或者wittig-horner 反应得到式a3所示化合物,更优选还包括使式a1所示化合物进行消除反应得到式a2 所示化合物,更优选还包括使式sm所示化合物进行氧化反应得到式a1所示化合物。
[0128]
本发明还提供一种胆酸中间体a5,包括采用如上所述胆酸中间体a5的制备方法制
备得到。
[0129]
本发明还提供一种胆酸的合成方法,所述胆酸的分子式为式ca所示,所述合成路线包括采用如上所述的胆酸中间体a5制备式a6所示化合物,再以所述式a6所示化合物制备式a7所示化合物,再以所述式a7所示化合物制备式a8所示化合物,再以所述式 a8所示化合物制备所述胆酸;优选的,在溶剂中,使式a5所示化合物在酸性条件下进行水解反应得到式a6所示化合物,再在溶剂中,使所述式a6所示化合物在催化剂钯碳的催化下进行氢化还原反应得到式a7所示化合物,再在溶剂中,使所述式a7所示化合物在碱存在的条件,进行水解反应,得到式a8所示化合物,再在溶剂存在下,使所述式 a8所示化合物与还原剂三叔丁氧基氢化铝锂进行选择性还原反应,得到所述胆酸。
[0130]
本发明还提供一种胆酸中间体a4,所述胆酸中间体a4的结构式为式a4所示。
[0131]
本发明还提供一种胆酸中间体a4的制备方法,所述胆酸中间体a4的结构式为式a4 所示,所述制备方法包括以下步骤:以甲苯为溶剂,以乙二醇为缩酮反应试剂,以对甲苯磺酸为催化剂,将式a3所示化合物与甲苯、乙二醇和对甲苯磺酸混合后,使体系升温至回流状态进行反应,反应完成后得到反应产物,所述反应产物经后处理后得到胆酸中间体a4;或者,以乙二醇为缩酮反应试剂,以原甲酸三乙酯为缩酮脱水剂,以对甲苯磺酸为催化剂,使a3所示化合物在35~80℃下进行反应,反应完成后得到反应产物,所述反应产物经后处理后得到胆酸中间体a4。
[0132]
在该胆酸中间体a4的制备方法中,优选包括使式a2所示化合物进行wittig或者 wittig-horner反应得到式a3所示化合物,更优选还包括使式a1所示化合物进行消除反应得到式a2所示化合物,更优选还包括使式sm所示化合物进行氧化反应得到式a1所示化合物。
[0133]
本发明还提供一种胆酸中间体a4,包括采用如上所述胆酸中间体a4的制备方法制备得到。
[0134]
本发明还提供一种胆酸的合成方法,所述胆酸的分子式为式ca所示,所述合成路线包括采用如上所述的胆酸中间体a4制备所述式a5所示化合物,再以所述式a5所示化合物制备式a6所示化合物,再以所述式a6所示化合物制备式a7所示化合物,再以所述式a7所示化合物制备式a8所示化合物,再以所述式a8所示化合物制备所述胆酸;优选的,在溶剂中,使a4所示化合物进行氧化反应得到式a5所示化合物,再在溶剂中,使式a5所示化合物在酸性条件下进行水解反应得到式a6所示化合物,再在溶剂中,使所述a6所示化合物在催化剂钯碳的催化下进行氢化还原反应得到式a7所示化合物,再在溶剂中,使所述式a7所示化合物在碱存在的条件,进行水解反应,得到式a8 所示化合物,再在溶剂存在下,使所述式a8所示化合物与还原剂三叔丁氧基氢化铝锂进行选择性还原反应,得到所述胆酸。
[0135]
本发明还提供一种胆酸中间体a3,所述胆酸中间体a3的结构式为式a3所示。
[0136]
本发明还提供一种胆酸中间体a3的制备方法,所述胆酸中间体a3的结构式为式a3 所示,所述制备方法包括以下步骤:进行wittig反应,以甲苯或四氢呋喃为溶剂,搅拌状态下加入式a2所示化合物和wittig试剂乙氧甲酰基亚甲基三苯基膦得到反应物体系,使所述反应物体系升温至回流状态反应预设时间直至反应完全,得到包括式a3所示化合物的反应产物,降至室温后,所述反应产物经后处理得到胆酸中间体a3;或者,进行 wittig-horner反应,以四氢呋喃为溶剂,使式a2所示化合物与碱和膦酰基乙酸三乙酯在 20~66℃下进行
反应,直至反应完全,得到包括胆酸中间体a3的反应产物,所述碱为氢化钠、叔丁醇钾、叔丁醇钠、甲醇钠、甲醇钾、乙醇钠、乙醇钾中的一种。
[0137]
在该胆酸中间体a3的制备方法中,优选还包括使式a1所示化合物进行消除反应得到式a2所示化合物,更优选还包括使式sm所示化合物进行氧化反应得到式a1所示化合物。
[0138]
本发明还提供一种胆酸中间体a3,包括采用如上所述胆酸中间体a3的制备方法制备得到。
[0139]
本发明还提供一种胆酸的合成方法,所述胆酸的分子式为式ca所示,所述合成路线包括采用如上所述的胆酸中间体a3制备式a4所示化合物,再以所述式a4所示化合物制备式a5所示化合物,再以所述式a5所示化合物制备a6所示化合物,再以所述a6所示化合物制备式a7所示化合物,再以所述式a7所示化合物制备所述式a8所示化合物,再以所述式a8所示化合物制备所述胆酸;优选的,在溶剂中,使a3所示化合物进行缩酮保护反应得到式a4所示化合物,再在溶剂中,使a4所示化合物进行氧化反应得到式a5所示化合物,再在溶剂中,使式a5所示化合物在酸性条件下进行水解反应得到式a6所示化合物,再在溶剂中,使所述a6所示化合物在催化剂钯碳的催化下进行氢化还原反应得到式a7所示化合物,再在溶剂中,使所述式a7所示化合物在碱存在的条件,进行水解反应,得到式a8所示化合物,再在溶剂存在下,使所述式a8所示化合物与还原剂三叔丁氧基氢化铝锂进行选择性还原反应,得到所述胆酸。
[0140]
本发明还提供一种胆酸中间体a2,所述胆酸中间体a2的结构式为式a2所示。
[0141]
本发明还提供一种胆酸中间体a2的制备方法,所述胆酸中间体a2的结构式为式a2 所示,所述制备方法包括以下步骤:以水为溶剂,使a1所示化合物与浓硫酸和冰醋酸在 20~50℃下进行反应直至反应完全,得到包括式a2所示化合物的反应产物,所述反应产物经后处理得到胆酸中间体a2;或者,以二氯甲烷或者三氯甲烷为溶剂,使式a1所示化合物与醋酸酐和浓硫酸在30~40℃下反应3~5h,直至反应完全,得到包括式a2所示化合物的反应产物。
[0142]
在该胆酸中间体a2的制备方法中,优选使式sm所示化合物进行氧化反应得到式 a1所示化合物。
[0143]
本发明还提供一种胆酸中间体a2,包括采用如上所述胆酸中间体a2的制备方法制备得到。
[0144]
本发明还提供一种胆酸的合成方法,所述胆酸的分子式为式ca所示,所述合成路线包括采用如上所述的胆酸中间体a2制备式a3所示化合物,再以所述式a3所示化合物制备式a4所示化合物,再以所述式a4所示化合物制备式a5所示化合物,再以所述式 a5所示化合物制备式a6所示化合物,再以所述式a6所示化合物制备式a7所示化合物,再以所述式a7所示化合物制备式a8所示化合物,再以所述式a8所示化合物制备所述胆酸;优选的,在溶剂中,使a2所示化合物进行wittig或者wittig-horner反应得到式a3所示化合物,再在溶剂中,使a3所示化合物进行缩酮保护反应得到式a4所示化合物,再在溶剂中,使a4所示化合物进行氧化反应得到式a5所示化合物,再在溶剂中,使式a5所示化合物在酸性条件下进行水解反应得到式a6所示化合物,再在溶剂中,使所述a6所示化合物在催化剂钯碳的催化下进行氢化还原反应得到式a7所示化合物,再在溶剂中,使所述式a7所示化合物在碱存在的条件,进行水解反应,得到式a8 所示化合物,再在溶剂存在下,使所述式a8所示化合物与还原
所示),将体系降温至8℃,控温滴加100.0ml琼斯试剂,保温搅拌6h,tlc检测反应至完全。加入100.0ml异丙醇淬灭反应,将体系缓慢加入到5000.0ml冰水中水析,降温至 6℃,过滤,干燥得到95.1g式a1所示化合物。
[0156]
分析数据为:
[0157]1hnmr(400mhz,cdcl3):δ9.55(s,1h),5.71(s,1h),3.51(s,1h-9-oh), 2.99-1.12(m,20h),1.37(s,3h-19-ch3),1.14(d,3h-21-ch3),1.06(s,3h-18
‑ꢀ
ch3)。
[0158]
实施例3~4合成式a2所示化合物
[0159]
合成路线如下式所示:
[0160][0161]
实施例3
[0162]
在洁净干燥的反应瓶中加入48.0ml水,冰水浴降温至8℃,搅拌下控温t≤40℃,滴加351.0g浓硫酸,加入96.0ml冰醋酸和95.0g式a1所示化合物,将体系控温至37℃反应2.5h,tlc检测反应至完全。将体系加入到1500.0ml冰水浴中,加完后搅拌30分钟以上,过滤,用水淋洗至中性,将滤饼用400.0ml二氯甲烷溶解,分液,收集有机层,减压浓缩,甲醇置换,最终浓缩至粘稠状态,过滤,滤饼干燥至合格,得到87.0g式a2所示化合物。
[0163]
实施例4
[0164]
在洁净干燥的反应瓶中加入570ml二氯甲烷,搅拌下加入95.0g式a1所示化合物,降温至6℃,滴加95.0ml醋酸酐,加完后搅拌15分钟,滴加38.0ml浓硫酸,滴加完成后升温至35℃,保温搅拌4h,tlc检测反应至完全。在体系中加入95.0ml水淬灭,分液,收集有机相,减压浓缩,甲醇置换,最终浓缩至粘稠状态,过滤,滤饼干燥至合格,得到86.7g式a2所示化合物。
[0165]
分析数据为:
[0166]1hnmr(400mhz,cdcl3):δ9.55(s,1h),5.71(s,1h),5.23(m,1h),3.00
‑ꢀ
1.13(m,18h),1.38(s,3h-19-ch3),1.13(d,3h-21-ch3),1.06(s,3h-18
‑ꢀ
ch3)。
[0167]
实施例5~7合成式a3所示化合物
[0168]
合成路线如下式所示:
[0169][0170]
实施例5
[0171]
在洁净干燥的反应瓶中加入1050.0ml甲苯,搅拌下加入85.0g式a2所示化合物和 173.0g乙氧甲酰基亚甲基三苯基膦,将体系升温至回流状态反应12h,tlc检测反应至完全,降至室温。在体系中加入氯化锌溶液(355.0ml水+210.0g氯化锌),于28℃下保温 12h进行
沉淀反应,抽滤,将沉淀物过滤,滤液进行分液,再次用氯化锌溶液(142.0ml 水+85.0g氯化锌)重复操作一次。过滤后分液,收集有机相,减压浓干,过滤,干燥得到94.3g式a3所示化合物。
[0172]
实施例6
[0173]
在洁净干燥的反应瓶中加入170.0ml四氢呋喃,搅拌下加入18.0g氢化钠,有气泡冒出,氮气保护,室温下滴加85.0ml膦酰基乙酸三乙酯,搅拌25分钟,滴加85.0g式a2 所示化合物和850.0ml四氢呋喃混合液,滴加完成后控温40℃反应2h,tlc检测反应至完全,室温下用6n盐酸调节ph=6.5,减压浓干四氢呋喃,加入200.0ml水水析,降温,过滤,干燥得到95.0g式a3所示化合物。
[0174]
实施例7
[0175]
在洁净干燥的反应瓶中加入170.0ml四氢呋喃,搅拌下加入18.0g氢化钠,有气泡冒出,氮气保护,室温下滴加85.0ml膦酰基乙酸三乙酯,搅拌25分钟,滴加85.0g式a2 所示化合物和850.0ml四氢呋喃混合液,滴加完成后控温40℃反应2h,tlc检测反应至完全,室温下用6n盐酸调节ph=6.5,减压浓干四氢呋喃,加入200.0ml水水析,降温,过滤,干燥得到95.0g式a3所示化合物。
[0176]
分析数据为:
[0177]1hnmr(400mhz,cdcl3):δ6.82(dd,j=15.3,9.0hz,1h),5.85(d, 1h),5.72(d,j=15.6hz,1h),5.25(m,1h-11-ch),4.26-4.09(m,2h), 3.05-2.86(m,2h),2.40-2.26(m,1h-20-ch),2.10-1.12(m,18h),1.37(s,3h
‑ꢀ
19-ch3),1.12(d,3h-21-ch3),1.06(s,3h-18-ch3)
[0178]
实施例8~9合成式a4所示化合物
[0179]
合成路线如下式所示:
[0180][0181]
实施例8
[0182]
在洁净干燥的反应瓶中加入640.0ml乙二醇、143.0ml原甲酸三乙酯和1.4g对甲苯磺酸,搅拌下加入93.0g胆酸中间体a3,将体系升温至45℃反应7h,tlc检测反应至完全,降温至室温,加入三乙胺调节ph至7.2。在体系中加入3500.0ml水水析,降温,过滤。滤饼用甲醇重结晶,降温,过滤,干燥得到84.6g式a4所示化合物。
[0183]
实施例9
[0184]
在洁净干燥的反应瓶中930.0ml甲苯、186.0ml乙二醇、0.93gpts和93.0g胆酸中间体a3,将体系升温至回流状态反应25h,tlc检测反应至完全。降温,室温下加入少量三乙胺调节ph=7.2,加入370ml水水洗,共洗2次,收集有机相,将甲苯层减压浓缩至粘稠状态,加入370.0ml石油醚,降温至6℃,过滤,干燥得到81.9g式a4所示化合物。
[0185]
分析数据为:
[0186]1hnmr(400mhz,cdcl3):δ6.82(dd,j=15.6,8.9hz,1h),5.72(d,j= 15.6hz,1h),
5.23(m,1h-11-ch),5.18(m,1h),4.18(q,j=7.1hz,2h), 3.97-3.90(m,4h),2.31-1.12(m,21h),1.37(s,3h-19-ch3),1.12(d,3h-21
‑ꢀ
ch3),1.06(s,3h-18-ch3)
[0187]
实施例10~13:合成式a5所示化合物
[0188]
合成路线如下式所示:
[0189][0190]
实施例10
[0191]
在洁净的反应瓶中加入1200.0ml丙酮和133.0ml水,搅拌下加入83.0g式a4所示化合物、83.0g氧化铬和60.0gnhpi,控温22℃,保温搅拌3h后,再加入83.0g氧化铬,保温搅拌23h,tlc检测反应至完全。加入72.0g异丙醇淬灭反应,将丙酮浓干,加入 500.0ml二氯甲烷,分液,有机相用水水洗一次,然后收集有机相。将处理的有机层减压浓缩,甲醇置换,降温,过滤,干燥得到64.4g式a5所示化合物。
[0192]
实施例11
[0193]
在洁净的反应瓶中加入1200.0ml丙酮和133.0ml水,搅拌下加入83.0g式a4所示化合物、100.0g氯铬酸吡啶翁和60.0gnhpi,控温22℃,保温搅拌5h后,再加入100.0g氯铬酸吡啶翁,保温搅拌23h,tlc检测反应至完全。加入80.0g异丙醇淬灭反应,将丙酮浓干,加入500.0ml二氯甲烷,分液,有机相用水水洗一次,然后收集有机相。将处理的有机层减压浓缩,甲醇置换,降温,过滤,干燥得到63.9g式a5所示化合物。
[0194]
实施例12
[0195]
在洁净的反应瓶中加入1600.0ml乙腈和133.0ml水,搅拌下加入83.0g式a4所示化合物、83.0g氧化铬和60.0gnhpi,控温22℃,保温搅拌4h后,再加入83.0g氧化铬,保温搅拌23h,tlc检测反应至完全。加入72.0g异丙醇淬灭反应,将丙酮浓干,加入 500.0ml二氯甲烷,分液,有机相用水水洗一次,然后收集有机相。将处理的有机层减压浓缩,甲醇置换,降温,过滤,干燥得到63.8g式a5所示化合物。
[0196]
实施例13
[0197]
在洁净的反应瓶中加入1600.0ml乙腈和133.0ml水,搅拌下加入83.0g式a4所示化合物、100.0g氯铬酸吡啶翁和60.0gnhpi,控温22℃,保温搅拌3h后,再加入100.0g氯铬酸吡啶翁,保温搅拌23h,tlc检测反应至完全。加入80.0g异丙醇淬灭反应,将丙酮浓干,加入500.0ml二氯甲烷,分液,有机相用水水洗一次,然后收集有机相。将处理的有机层减压浓缩,甲醇置换,降温,过滤,干燥得到63.3g式a5所示化合物。
[0198]
分析数据为:
[0199]1hnmr(400mhz,cdcl3):δ6.82(dd,j=15.6,9.0hz,1h),5.99(s, 1h),5.90-5.70(m,2h),4.16(q,j=7.1hz,2h),3.97-3.90(m,4h),3.04 (m,1h),2.31-1.12(m,16h),1.37(s,3h-19-ch3),1.12(d,3h-21-ch3), 1.06(s,3h-18-ch3)。
[0200]
实施例14:合成式a6所示化合物
[0201]
合成路线如下式所示:
[0202][0203]
在洁净的反应瓶中加入630.0ml甲醇、63.0ml水和38.0ml浓盐酸,搅拌下加入63.0g 式a5所示化合物,将体系升温至55℃反应4h,tlc检测反应至完全。将体系降温至 25℃,用饱和碳酸氢钠溶液调节ph至6.5,过滤。干燥得到55.1g式a6所示化合物。
[0204]
分析数据为:
[0205]1hnmr(400mhz,cdcl3):δ6.82(dd,j=15.6,9.0hz,1h),5.99(s, 1h),5.90-5.70(m,2h),4.16(q,j=7.1hz,2h),3.20-2.80(m,5h),2.35 (m,1h),2.21-1.13(m,11h),1.37(s,3h-19-ch3),1.13(d,3h-21-ch3), 1.04(s,3h-18-ch3)。
[0206]
实施例15~17:合成式a7所示化合物
[0207]
合成路线如下式所示:
[0208][0209]
实施例15
[0210]
在洁净干燥的高压罐中加入270.0ml无水乙醇和270.0ml吡啶,搅拌下加入54.0g式 a6所示化合物和5.4g10%钯碳,在35℃用氢气加压至0.42mpa反应10h,tlc检测反应至完全。将体系过滤除钯碳,然后减压浓缩,用水置换,水析出料,少量水淋洗,滤饼干燥至合格,得到50.6g式a7所示化合物,其中,a7纯度:5α-h异构体纯度=89.8: 10.2。
[0211]
实施例16
[0212]
在洁净干燥的高压罐中加入270.0ml无水乙醇、216.0ml吡啶和54.0ml1-甲基咪唑,搅拌下加入54.0g式a6所示化合物和5.4g10%钯碳,在35℃用氢气加压至0.42mpa反应 10h,tlc检测反应至完全。将体系过滤除钯碳,然后减压浓缩,用水置换,水析出料,少量水淋洗,滤饼干燥至合格,得到51.3g式a7所示化合物,其中,a7纯度:5α-h异构体纯度=92.7:7.3。
[0213]
实施例17
[0214]
在洁净干燥的高压罐中加入270.0ml无水乙醇、270.0ml吡啶和5.4gdmap,搅拌下加入54.0g式a6所示化合物和5.4g10%钯碳,在35℃用氢气加压至0.42mpa反应10h, tlc检测反应至完全。将体系过滤除钯碳,然后水析出料,少量水淋洗,滤饼干燥至合格,得到50.9g式a7所示化合物,其中,a7纯度:5α-h异构体纯度=98.3:1.7。
1.06(s,3h,18-ch3);0.84(d,3h,j=6.5hz,21-ch3)。
[0229]
实施例23:合成胆酸
[0230]
胆酸的结构式为式ca所示,合成路线如下式所示:
[0231][0232]
在洁净干燥的反应瓶中加入700.0ml四氢呋喃和45.0g式a8所示化合物,氮气保护下降温至3℃,分批缓慢加入130.0g三叔丁氧基氢化铝锂,加完后升温至25℃反应11h,tlc检测反应至完全,缓慢滴加1n盐酸,调节ph=2.5,减压浓干四氢呋喃,水析出料,少量水淋洗,滤饼用乙酸乙酯溶清后分液,重结晶精制,得到28.1g目标产物胆酸。
[0233]
分析数据为:
[0234]
1h-nmr((d5)pyridine,500mhz)::4.25(s,h-c(12));4.09(s,h-c (7));3.78
–
3.73(m,h-c(3));3.11(q,j=13.0,h-c(8));2.92(dt, j=12.0,4.2,h-c(9));2.79
–
2.73(m,h-c(14));2.67
–
2.61(m,h-c (23a));2.56
–
2.50(m,h-c(23b));2.39
–
2.34(m,h-c(17));2.15
–ꢀ
2.13(m,ch2(22));2.13
–
2.11(m,h-c(4a));2.07
–
2.00(m,h-c (15a));1.94
–
1.91(m,h-c(1a,2a));1.88
–
1.86(m,h-c(11a));1.68
–ꢀ
1.67(m,ch2(6));1.67
–
1.62(m,h-c(20,4b,2b,11b));1.51
–
1.49(m,h-c (5));1.44
–
1.37(m,ch2(16));1.23(d,j=5.5,me(21));1.21
–
1.19 (m,h-c(15b);1.09
–
1.03(m,h-c(1b));1.00(s,me(19));0.81(s,me (18))。
[0235]
以上内容是结合具体的优选实施方式对本发明作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演和替换,都应当视为属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1