电子薄壁制件用芳香族液晶聚合物及其组合物的制作方法
1.本发明涉及热稳定性高、流动性良好的芳香族液晶聚合物及其组合物,由该液晶聚合物及其组合物成型的电子薄壁制件翘曲变形得到充分抑制。
背景技术:
2.液晶聚合物(lcp)材料具有优异的机械性能、尺寸稳定性、电性能、耐化学药品性、阻燃性、良好的加工性、耐热性、较低的热膨胀系数等特点。近年来,随着电子产业的快速发展,特别是5g通讯产业的迅猛发展,更是将电子产业的发展推向了新高度。电子部件日益向轻、小、薄的方向发展,为了适应这样的应用扩展和变化,需要开发具备高热稳定性的液晶聚合物材料,以满足电子领域的无铅焊锡要求;同时,液晶聚合物材料还要具备较好的流动性,使产品在成型加工过程中产生更低的成型应力,确保产品的尺寸稳定性,另外较低的成型应力还可以防止在成型后及回流焊加热过程中产生翘曲变形,导致与基板的焊接不良。
3.现有技术已经进行多种尝试通过降低液晶聚合物熔体粘度来改善其流动性。专利us09051514b2通过将添加芳族酰胺低聚物改变分子间聚合物链相互作用而降低聚合物的熔融粘度,专利jp33012988通过向液晶聚合物加入对羟基苯甲酸的低聚物的方法改善流动性,通过以上方法液晶聚合物的流动性虽得到明显改善,但其在无铅焊接和其他制造过程中易起泡,热稳定性能欠佳。
技术实现要素:
4.本发明的目的是解决上述现有技术存在的问题,提供一种即使是精密复杂的电子薄壁制件也能够成型为流动性良好、热稳定性优异、翘曲变形得到充分抑制的成型品用芳香族液晶材料。
5.本发明人为了实现上述目的,提供性能得到良好平衡并且适用于成型电子薄壁制件的材料,经过深入探索和研究发现,通过使用包含特定结构、含量为特定范围的单体聚合而成的芳香族液晶聚合物及其组合物,可以解决上述技术问题。具体而言,本发明提供以下方案。
6.一种芳香族液晶聚合物,其由下述结构单元(ⅰ)、(ⅱ)、(ⅲ)、(ⅳ)作为必须的构成成分:
7.结构单元(ⅰ)相对于全部结构单元的含量为46~70mol%;
8.结构单元(ⅱ)相对于全部结构单元的含量为14.5~24mol%;
9.结构单元(ⅲ)相对于全部结构单元的含量为0.5~3mol%;
10.结构单元(ⅳ)相对于全部结构单元的含量为15~27mol%;
11.结构单元(ⅰ)、(ⅱ)、(ⅲ)、(ⅳ)的总计含量为100mol%。
12.(ⅰ)
13.14.(ⅱ)
[0015][0016]
(ⅲ)
[0017][0018]
(ⅳ)
[0019][0020]
结构单元(ⅰ)的含量低于46mol%时,形成的聚合物易凝固黏连在反应釜壁,不能顺利从反应釜排出;结构单元(ⅰ)的含量超过70mol%时,聚合物的熔点降低,热稳定性能下降。从熔点和聚合的角度出发,结构单元(ⅰ)的含量应当控制在46~70mol%范围内。
[0021]
结构单元(ⅱ)的含量低于14.5mol%时,聚合物熔点降低,热稳定性能下降;结构单元(ⅱ)的含量超过24mol%时,聚合物易凝固黏连在反应釜壁,不能顺利排出反应釜。从熔点和聚合的角度出发,结构单元(ⅱ)的含量应当控制在14.5~24mol%范围内。
[0022]
结构单元(ⅲ)含有邻二氮杂菲骨架,赋予液晶聚合物分子长链适当弯曲结构,打破分子链规整性,降低液晶聚合物各向异性,提高其流动性;另外,其结构单元中含有吡啶环,与苯环相比,吡啶环上的氮原子使其邻、对位上的电子云密度降低,导致吡啶环上处于邻位的基团键能降低,使得聚合物热稳定性下降。因此,从流动性和热稳定性角度出发,结构单元(ⅲ)的含量应当控制在0.5~3mol%的范围。
[0023]
结构单元(ⅳ)的含量低于15mol%时,聚合物热稳定性下降,随着结构单元(ⅳ)含量的增加,聚合物分子量有所增加,流动性有变差的趋势,为了保持流动性和热稳定性的平衡,结构单元(ⅳ)的含量不超过27mol%。
[0024]
在上述芳香族液晶聚合物中,优选结构单元(ⅳ)的含量为(ⅱ)和(ⅲ)含量之和。
[0025]
在上述芳香族液晶聚合物中,优选在芳香族液晶聚合物熔点tm~[(tm+20)]℃温度范围下,剪切速率1000s-1
下测定的熔体粘度为30~60pa.s。
[0026]
在上述芳香族液晶聚合物中,优选液晶聚合物的熔点tm在300℃以上。
[0027]
一般情况下,在液晶聚合物熔点以上其粘度明显降低,流动性增加,但后期成型加工过程中易出现热劣化及对加工设备热能力要求高。因此,为了使热性能和加工条件达到一个相对平衡的状态,优选液晶聚合物的熔点在300~360℃。
[0028]
在上述芳香族液晶聚合物中,优选液晶聚合物的重均分子量为15000以上。
[0029]
进一步优选,液晶聚合物的重均分子量在20000-45000。
[0030]
在上述芳香族液晶聚合物中,优选10mg所述液晶聚合物在360℃保持20min产生气体量不超过12000ppm。
[0031]
在上述芳香族液晶聚合物中,优选结构单元(ⅰ)衍生自对羟基苯甲酸、结构单元
(ⅱ)衍生自2,6-萘二甲酸、结构单元(ⅲ)衍生自1,10-邻二氮杂菲-2,9-二甲酸、结构单元(ⅳ)衍生自对苯二酚。
[0032]
本发明芳香族液晶聚合物优选熔融固相缩聚方法进行聚合,具体可以为:将各单体原料、乙酸酐、催化剂投入哈氏合金聚合釜后于130-150℃下保持2-6h;以0.2-1.0℃/min的速度升温至300-330℃,保温1-2h;向聚合釜中冲入0.1-1.0mpa氮气,预聚物经过直径2-4mm的8-10孔放料阀门放出,粉碎,过20-30目筛,经130-150℃干燥2-3h后制得预聚物;将制得的预聚物在氮气保护下,于190-300℃的旋转窑中固相缩聚12-36h,制得液晶聚合物。
[0033]
上述聚合催化剂可以为乙酸镁、乙酸钾、乙酸钠、三氧化二锑、钛酸四丁酯、硫酸亚锡、4-二甲氨基嘧啶,优选4-二甲氨基嘧啶;催化剂添加量为所有单体总重量的30-500ppm。
[0034]
上述聚合乙酸酐添加量优选所有单体羟基总摩尔数的1.0-3.0倍。
[0035]
本发明的第二个目的是提供一种液晶聚合物组合物,其含有上述芳香族液晶聚合物。
[0036]
在上述液晶聚合物组合物中,可以根据使用目的添加各种无机和有机填充剂。相对于芳香族液晶聚合物以100重量份计,无机和有机填充剂的添加量为20~60重量份。
[0037]
在上述液晶聚合物组合物中,可以在不损害本发明效果的范围内添加的无机填充剂有玻璃纤维、碳纤维、碳纳米管、云母粉、滑石粉、硅微粉、硅酸钙、碳酸钙、氧化钛等,其中以玻璃纤维为优选,玻璃纤维的平均长度为1-30μm,平均直径为0.5-5μm。
[0038]
本发明以具有良好热稳定性、流动性、低翘曲的液晶聚合物为基体,添加玻璃纤维改性,获得的组合物在具有良好热稳定性、流动性、低翘曲的同时,机械强度得到进一步改善。
[0039]
在上述液晶聚合物组合物中,可以在不损害本发明效果的范围内添加par、pet、pbt、pps、pi、ptfe、ppo等有机物。
[0040]
本发明的再一个目的是提供一种液晶聚合物电子薄壁制件,其由液晶聚合物或其组合物成型得到。
[0041]
本发明的液晶聚合物电子薄壁制件的制造方法没有特别限定,可以为注塑成型、挤出成型、压缩成型、气体注射成型等方法。
[0042]
本发明的芳香族液晶聚合物及其组合物可用于cpu风扇、薄壁型电子连接器、线圈架等,但不限于此。
[0043]
与现有技术相比,本发明具有如下有益效果:
[0044]
本发明能够提供成型为热稳定性高、流动性好的且翘曲变形小的成型品的芳香族液晶聚合物及包含该芳香族液晶聚合物的组合物。
具体实施方式
[0045]
以下结合实施例对本发明进一步举例说明,在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围包括但不限于以下特定的具体实施方案。一般地,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。以下实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
[0046]
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端
点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
[0047]
实施例1
[0048]
将相对于全部结构单元含量为60mol%的对羟基苯甲酸、20mol%的6-羟基-2-萘甲酸、1.0mol%1,10-邻二氮杂菲-2,9-二甲酸、19mol%对苯二酚、占所有单体羟基总摩尔数1.5倍的乙酸酐、占所有单体总重量100ppm的4-二甲氨基嘧啶投至哈氏合金聚合釜中后于135℃下保持3h;以0.5℃/min的速度升温至320℃,保温2h;向聚合釜中冲入0.5mpa氮气,预聚物经过直径2mm的8孔放料阀门放出,粉碎,过20目筛,经140℃干燥3h后制得预聚物;将制得的预聚物在氮气保护下,于240℃的旋转窑中固相缩聚24h,制得液晶聚合物。
[0049]
对实施例1的芳香族液晶聚合物按照如下方法进行熔点、熔融粘度、重均分子量、热稳定性、翘曲变形进行测试评价,评价结果详见表1。
[0050]
1、熔点
[0051]
使用差示扫描量热仪dsc-500c,从室温开始以15℃/min的升温速率升至能观测到的吸热峰温度(tm1)后,以(tm1+30)℃的温度保持2min后,以15℃/min的降温速率降至室温,再次以15℃/min的升温速率升至可观测的吸热峰温度tm2,该温度即为液晶聚合物的熔点。
[0052]
2、熔融粘度
[0053]
依据gb/t25278-2010测试方法,以高于液晶聚合物熔点0~20℃的温度,使用内径1mm、长度20mm的节流孔,以剪切速率1000s-1
,测定液晶聚合物的熔融粘度。
[0054]
3、重均分子量
[0055]
使用重量比为35/65的五氟苯酚/氯仿作为混合溶剂,将液晶聚合物溶解于混合溶剂,形成浓度为0.03-0.06重量/体积%溶液,待溶解完毕,提取上清液,采用凝胶渗透色谱分析仪器进行测定,通过聚苯乙烯进行换算得到液晶聚合物的重均分子量。
[0056]
4、热稳定性
[0057]
使用热重/差热综合分析仪tg/dta-6300,取10mg芳香族液晶聚合物在氮气气流下、以360℃保持20min时重量减少作为产生气体量。若产生气体量低于12000ppm,则热稳定性视为良好。
[0058]
5、翘曲度
[0059]
将干燥好的液晶聚合物在注射成型机上注塑成直径为64mm、厚度为0.5mm的盘状样品;然后,将获得的样品放在平板上,将盘的外周取作参考平面,并且将最远离平板的部分取作门部分,使用测微计测量从参考平面到门部分的高度,检查位移。所得位移数值作为成型制品的翘曲度。
[0060]
实施例2
[0061]
将与实施例1同等投入比例的原料单体、占所有单体羟基总摩尔数1.0倍的乙酸酐、占所有单体总重量30ppm的4-二甲氨基嘧啶投至哈氏合金聚合釜中后于130℃下保持6h;以0.2℃/min的速度升温至300℃,保温2h;向聚合釜中冲入0.1mpa氮气,预聚物经过直
径3mm的9孔放料阀门放出,粉碎,过20目筛,经130℃干燥3h后制得预聚物;将制得的预聚物在氮气保护下,于300℃的旋转窑中固相缩聚12h,制得液晶聚合物。与实施例1进行同样的性能测试评价,评价结果详见表1。
[0062]
实施例3
[0063]
将与实施例1同等投入比例的原料单体、占所有单体羟基总摩尔数3.0倍的乙酸酐、占所有单体总重量500ppm的4-二甲氨基嘧啶投至哈氏合金聚合釜中后于150℃下保持2h;以1.0℃/min的速度升温至330℃,保温1h;向聚合釜中冲入1.0mpa氮气,预聚物经过直径4mm的10孔放料阀门放出,粉碎,过30目筛,经150℃干燥2h后制得预聚物;将制得的预聚物在氮气保护下,于190℃的旋转窑中固相缩聚36h,制得液晶聚合物。与实施例1进行同样的性能测试评价,评价结果详见表1。
[0064]
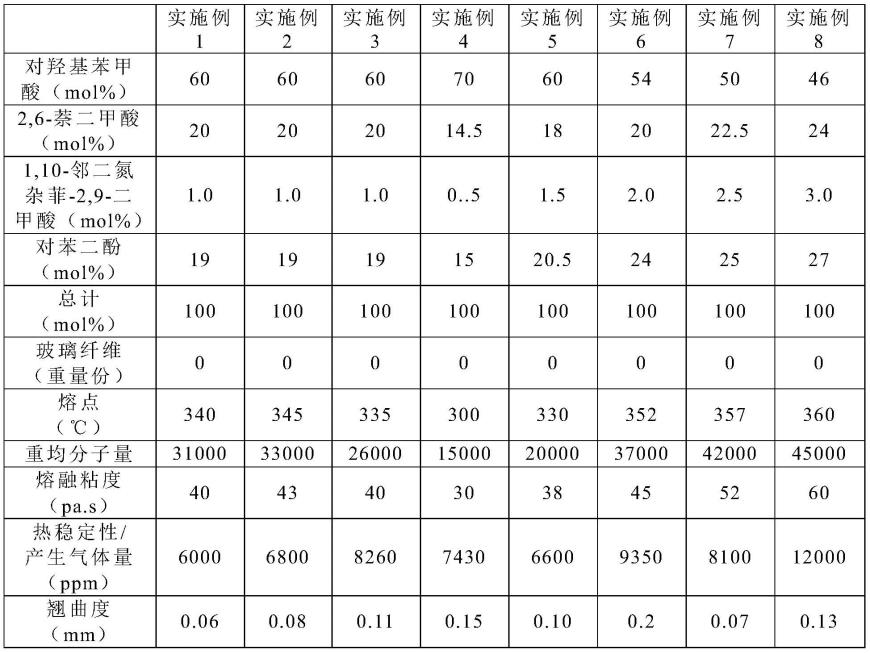
实施例4-8及对比例1-8的原料种类及投入比例如下表1和表2所示,另外其性能评价方法同实施例1,具体性能评价结果见表1和表2。需要特别说明的是:
①
、对比例4在聚合过程中出现凝固现象,无法顺利排出反应釜;
②
、对比例8与实施例1的区别仅在于,聚合催化剂选用金属盐催化剂乙酸钾。未特别说明的实施例和对比例制备过程皆同实施例1。
[0065]
表(1)
[0066][0067]
表(2)
[0068][0069][0070]
如上表1和表2所示,与未使用本发明技术方案的对比例1-8相比,应用了本发明技术方案的实施例1-8具有高的热稳定性、良好的流动性、低翘曲等优异性能,符合电子薄壁制件对材料的要求,具有广泛的工业实用性。
[0071]
以上所述,对于本领域技术普通技术人员而言,可以根据本发明的技术方案和技术构思作出各种相应的改变和变形,凡是依据本发明的技术实质或原料组分或含量对以上实施方式所作的任何细微修改、等同变化与修饰,均属于本发明技术方案保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1