激酶基因在调控丝状真菌菌丝形态中的应用
1.本发明涉及基因工程技术领域,具体涉及激酶基因在调控丝状真菌菌丝形态中的应用。利用基因调控黑曲霉的菌丝结构呈简单化,以及在黑曲霉总蛋白分泌方面的用途,为丝状真菌的形态调控以及分子遗传学操作领域提供可靠的方法和依据。
背景技术:
2.丝状真菌作为一种高效的细胞工厂,被广泛应用于生物制造领域。它可以用来生产工业酶制剂(蛋白酶、淀粉酶、纤维素酶、脂肪酶)、多糖、有机酸、抗生素、生物膜等初级或次级代谢产物。尤其具备表达天然或重组蛋白的能力。例如,黑曲霉(aspergillus niger)可商业化生产柠檬酸和葡萄糖氧化酶等。丝状真菌被用作高效的细胞工厂,与其独特的形态结构密切相关。
3.丝状真菌的主要特征是菌丝结构,例如,典型的丝状真菌黑曲霉的菌丝结构呈管状菌丝形态,即一个菌丝包括长长的杆状主菌丝体、分支菌丝体,分生孢子聚集体。在许多丝状真菌中,个体的菌丝可以与同一个体的其他菌丝融合形成菌丝网络。在深层培养中,宏观形态从分散的菌丝体,到菌丝的簇状聚集体,再到直径几毫米的近球形紧密菌丝颗粒。分散的菌丝可以增加某些酸(富马酸)、蛋白质(淀粉酶、新果糖转移酶、植酸酶)和次生代谢物(青霉素)的产生。然而,颗粒状宏观形态有利于某些分子的产生,包括柠檬酸、糖化酶或聚半乳糖醛酸酶。菌丝生长为底物的定植、水解酶的分泌、营养物质的同化、形态发生的调节和环境信号的识别提供了手段。菌丝的生长和分化是一个复杂的过程,需要控制细胞壁的合成、极化的囊泡运输、胞吞作用、吞噬作用、膨压、细胞器的定位以及细胞质的迁移和融合。此外,丝状真菌与其他多细胞生物有所不同,非同核细胞的丝状真菌,其细胞核可以通过隔膜孔在整个菌丝体网络中自由移动,从而导致丝状真菌具备多核的结构。丝状真菌的特殊结构增加了对其进行遗传改造的难度,一方面遗传物质较难穿过复杂的细胞网络以及细胞壁进入细胞内,另一方面较难获得比较纯的同核突变体。同时,丝状真菌复杂的菌丝结构不利于高通量技术的使用,增加了对其的遗传改造难度。因此,克服丝状真菌的复杂结构导致的遗传操作、高通量筛选的困难,有利于促进我们对丝状真菌的理解和在工业上的应用。综上所述,寻求一种简单的调控丝状真菌菌丝状结的方法对于丝状真菌的研究和应用十分重要。
技术实现要素:
4.本发明的目的在于克服现有技术的缺点与不足,提供一种激酶基因在调控丝状真菌菌丝形态中的应用。通过本发明提供的方法可以调控丝状真菌的菌丝结构呈简单的短杆状,为丝状真菌的遗传操作以及高通量筛提供了材料和依据。
5.本发明的另一目的在于提供一株黑曲霉突变株,可以提高菌株分泌总蛋白的能力,可以用于工业发酵领域。
6.本发明的目的通过下述技术方案实现:
7.激酶基因在调控丝状真菌菌丝形态中的应用,其中,所述的激酶基因为丝状真菌mapk途径和/或camp-pka途径上的激酶基因。
8.所述的mapk途径为细胞完整性途径,高渗透压甘油激酶途径,低压/外界激素刺激激酶途径,孢子形成激酶途径中的至少一种;优选的,所述的mapk途径为细胞完整性途径中的级联激酶途径。
9.所述的激酶基因为基因bck1、mkk2、mpka、stec、pkac中的至少一种;优选的,所述的激酶基因为基因mpka。
10.所述的基因bck1的核苷酸序列的fungidb数据库序列登记号为an02g06830,基因mkk2的核苷酸序列的fungidb数据库序列登记号为an18g03740,基因mpka的核苷酸序列的ncbi数据库序列登记号为xm 025594238.1,基因stec的核苷酸序列的ncbi数据库序列登记号为xm 025594636.1基因mpka的核苷酸序列的ncbi数据库序列登记号为xm 025603413.1。
11.所述的基因bck1编码的蛋白质的氨基酸序列的fungidb数据库序列登记号为an02g06830,基因mkk2编码的蛋白质的氨基酸序列的fungidb数据库序列登记号为an18g03740,基因mpka编码的蛋白质的氨基酸序列的fungidb数据库序列登记号为an01g09520,基因stec编码的蛋白质的氨基酸序列的fungidb数据库序列登记号为an17g01280,基因pkac编码的蛋白质的氨基酸序列的fungidb数据库序列登记号为an02g04270。
12.所述的丝状真菌为黑曲霉,米曲霉,土曲霉,构巢曲霉,烟曲霉中的至少一种;优选的,所述的丝状真菌为黑曲霉;更优选为黑曲霉(aspergillus niger cbs513.88)。
13.所述的激酶基因被敲除或沉默后,丝状真菌的菌丝结构呈短杆状,分支少,极化生长周期短,不产分生孢子等表型出现,且分泌总蛋白的能力变强。
14.所述的敲除或沉默为使用包括crispr、sirna、碱基突变等技术对激酶基因的核苷酸序列进行敲除或编辑/突变/沉默,使激酶基因不表达;优选为使用crispr技术进行敲除或沉默;更优选为使用rnp基因编辑体系敲除或沉默。
15.本发明所述的应用,具体包括如下步骤:
16.(1)制备丝状真菌的原生质体;
17.(2)敲除或沉默激酶基因;
18.(3)在筛选平板上筛选,得到菌丝形态改变的丝状真菌。
19.所述的敲除或沉默为使用rnp基因编辑体系,具体步骤如下:
20.(a)根据需要敲除或沉默的激酶基因设计sgrna和修复片段;
21.(b)将sgrna与cas9酶组装,孵育得到rnp基因编辑体系;
22.(c)在丝状真菌原生质体中加入sgrna和修复片段,孵育,实现激酶基因的敲除或沉默。
23.所述的sgrna,是将启动子、spacer rna与sgrna骨架按顺序组装得到的。
24.所述的启动子为t7或u6启动子中的至少一种;优选为t7启动子。
25.当需要敲除的为基因mpka时,所述的spacer rna为其核苷酸序列中正向的第12个到第31个的序列。
26.当需要敲除的为基因stec时,所述的spacer rna为其核苷酸序列中正向的第28个到第47个的序列。
27.当需要敲除的为基因pkac时,所述的spacer rna为其核苷酸序列中正向的第262个到第281个的序列。
28.所述的修复片段包括筛选标签以及上下游同源臂,上下游同源臂的长度为39bp~1500bp。
29.所述的修复片段,是将spacer rna位点的前39bp上游同源序列、筛选标签和spacer rna位点的后39bp下游同源序列按顺序组装得到的。
30.所述的筛选标签为anpyrg筛选标签。
31.一种黑曲霉突变株,是黑曲霉敲除了上述的激酶基因得到的,提高了菌株分泌总蛋白的能力。
32.本发明相对于现有技术具有如下的优点及效果:
33.a.本发明中利用mapk途径上的基因来调控丝状真菌的菌丝结构,具备创新性,为丝状真菌的菌丝调控机制提供了理论依据。
34.b.本发明通过敲除mapk途径上的基因,可以导致复杂的菌丝呈现短杆状,分支少,极化生长周期短,不产分生孢子等表型。这些表型有利于丝状真菌的遗传操作以及高通量技术的实现。
35.c.本发明获得突变菌株不产孢子,同时总蛋白的分泌量增加,可以直接用于工业发酵领域。
36.d.本发明首次在丝状真菌中采用cas9和sgrna形成的核糖核蛋白复合物,简称rnp,组合修复片段的方法对丝状真菌中的基因进行编辑,对于高效编辑丝状真菌的基因具有参考意义。
附图说明
37.图1是rnp组合短修复片段的方法对丝状真菌中的基因进行敲除的示意图。
38.图2是rnp切割位点,修复片段同源重组作用示意图。
39.图3是实施例1中sgrna体外切割后鉴定的结果图;其中(a)为修复模板的pcr扩增后的核酸凝胶图;(b)为sgrna体外切割模板后的核酸凝胶图,(c)为激酶基因敲除菌株的pcr鉴定核酸凝胶图。
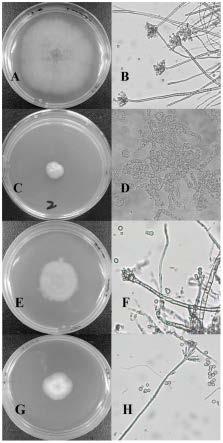
40.图4是实施例2中野生菌株及基因被敲除的突变体的菌落形态图和菌丝形态图;分别为:野生菌株wt的菌落图(a),野生菌株wt的菌丝形态图(b),丝状真菌mapk途径上的mpka基因被敲除的突变体菌落图(c),mpka基因被敲除的突变体菌丝形态图(d),丝状真菌mapk途径上的stec基因被敲除的突变体菌落图(e),stec基因被敲除的突变体菌丝形态图(f),camp-pka途径上pkac基因被敲除的突变体菌落图(g),pkac基因被敲除的突变体菌丝形态图(h)。
41.图5是野生菌株wt和激酶基因敲除菌株的菌落直径比较图。
42.图6是丝状真菌以及突变体的总蛋白分泌情况图。
具体实施方式
43.下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。
44.下面实施方案中若未注明具体试验条件,则通常按照常规试验条件或按照试剂公司所建议的试验条件。所使用的材料、试剂等,若无特殊说明,均为从商业途径得到的试剂和材料。
45.本发明所涉涉及的菌株、原材料、试剂、仪器:黑曲霉(aspergillus niger cbs513.88)在文献“董宏智.黑曲霉crispr基因编辑技术建立与内质网磷脂调控机制的研究[d].华南理工大学.”中公开,为本实验室保藏的材料;本发明涉及的化学试剂药品:脱脂奶粉、蜗牛酶、纤维素酶(北京鼎国昌盛生物技术公司),蛋白胨、酵母粉(英国oxoid公司),聚乙二醇(peg4000)、tris-base、山梨醇、无水氯化钙、氨苄青霉素、溶菌酶、尿嘧啶核苷、triton x-100(美国sigma公司),琼脂糖、限制性内切酶、dream taq green pcr master mix(美国thermo fisher公司),primerstar hs dnapolymerase、dna ligation kit、dna marker、fastap去磷酸化试剂盒、rnaiso plus、6
×
loading buffer、rnase free水、rna酶抑制剂、sybr premix ex taqtm(日本takara公司),gelred核酸染料(美国biotum公司),miracloth滤布(德国默克公司),pcr产物纯化试剂盒、质粒大量提取试剂盒(广州捷贝斯),infusion试剂盒(nebuilder hifi dnaassembly cloning kit)、t7体外转录试剂盒、cas9蛋白试剂盒、限制性内切酶(美国neb公司),其它化学试剂药品为国产或进口分析纯级别及以上;本发明所涉及的主要仪器设备:nanodrop1000生物分光光度计(美国thermo fisher公司),高压灭菌锅(mls-3750,日本sanyo公司),dna凝胶电泳仪(美国bio-rad公司),dna凝胶成像仪(美国carestream health公司),霉菌培养箱(shp-450d,上海森信实验仪器有限公司),恒温空气浴摇床(苏州培英实验仪器有限公司),小型台式离心机(德国eppendorf公司),超净工作台(sw-cj-1fd,苏州净化有限公司),ims-20制冰机(广州市深华生物技术有限公司),超纯水仪(上海领德仪器有限公司),电动组织研磨器(北京天根生化科技有限公司),高精度天平(德国赛多利斯公司),涡旋振荡器(美国scilogex公司),超声波清洗仪(宁波新芝仪器有限公司),真空泵(doa-p70-bn,美国pall公司)。
[0046]
实施例1
[0047]
本发明为了验证激酶基因在丝状真菌中的作用,设计实验敲除/沉默激酶基因后进行筛选和验证,可以根据实际需要选择sirna、编辑质粒、易错pcr等方法对激酶基因进行沉默或编辑,下述所使用的crispr编辑方法为本领域技术人员常用的技术方法,是发明人选择的较优的实验方法,所使用的序列及材料均为本领域技术人员可以根据需要从公开渠道获得的,但是本发明的实施方案并不限于这一种方法。
[0048]
黑曲霉rnp基因编辑体系的构建方法,包括如下步骤:
[0049]
(1)基因序列的获取。根据分析找到需要编辑的基因序列,基因库为ncbi或者fungidb。例如,通过fungidb基因库找到mpka基因的序列(id:an01g09520),stec基因的序列(id:an17g01280),以及pkac基因的序列(id:an02g04270)。
[0050]
(3)前间隔序列spacer rna的设计。在设计spacer rna之前,首先整理好要作用的基因序列,例如本发明涉及的具体基因mpka、stec、pkac。基因作用靶点设计在外显子上,同时基因的长度小于1000bp。选择的在线设计平台为:http://www.rgenome.net/cas-designer/。进入在线平台后,选择cas-designer。首先选择cas9种类,一般选择第一种spcas9 from streptococcus pyogenes(5端-ngg-3端)。选择物种和基因组。例如,要作用的基因来自黑曲霉,所以物种就选择黑曲霉,并在对用的框内输入整理好的基因序列,基因
序列小于1000bp。输入序列后,生成20nt spacer rna序列库,下载所有结果。将所有结果通过python程序进行分析,评分标准参考文献(john g doench et al.,rational design of highly active sgrnas for crispr-cas9
–
mediated gene inactivation.2014.nature biotechnology.),评分高的序列可以作为前间隔序列,即spacer rna序列。本发明中选择的spacer rna核酸序列如下所示。
[0051]
其中mpka基因的20nt spacer rna序列(gcagggtcgcaagatcttca)为ncbi数据库中序列id为xm 025594238.1的正向的第12个到第31个的序列,stec基因的20nt spacer rna序列(tgggttgcagacacacccaa)为ncbi数据库中序列id为xm 025594636.1的正向的第28个到第47个的序列,pkac基因的20nt spacer rna序列(ggagcagaaaaatcctccga)为ncbi数据库中序列id为xm 025603413.1的正向的第262个到第281个的序列。
[0052]
(4)前间隔序列spacer rna的体外靶向性的分析。为了验证设计的spacer rna具有靶向性,先进行体外切割分析。体外切割体系主要包括cas9酶,sgrna,被切割的dna模板,以及缓冲液。其中,cas9酶和缓冲液为商业渠道购买,sgrna需要自己转录,采用t7转录酶制备,t7转录体系为模板10μl、t7酶2μl、buffer 10μl、h2o 8μl,总体积共30μl,转录条件为37℃,16h。
[0053]
t7转录的spacer rna以及sgrna骨架的序列如下所示,sgrna是将t7转录序列、对应的spacer rna和sgrna骨架按顺序组装得到的,小写字母的序列为t7转录序列,大写字母未划线的序列为20nt的spacer rna序列,小写字母且划线的序列为sgrna骨架的序列。
[0054]
mpka-sgrna:
[0055]
taatacgactcactataggggcagggtcgcaagatcttcagttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgc;
[0056]
stec-sgrna:
[0057]
taatacgactcactatagggtgggttgcagacacacccaagttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgc;
[0058]
pkac-sgrna:
[0059]
taatacgactcactatagggggagcagaaaaatcctccgagttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgc。
[0060]
t7转录序列,以及sgrna骨架的序列的来源可以参考文献“yu leyi et al.,a special phenotype of aconidial aspergillus niger sh2 and its mechanism of formation via crispri.2022.journal of fungi.”[0061]
上述t7-spacer rna-sgrna骨架序列通过pcr获得。首先通过生物公司合成sgrna骨架序列,然后设计引物以生物公司合成的sgrna骨架序列作为模板扩增获得。设计的正向引物(t7-mpka spacer rna-sgrna骨架-f、t7-stec spacer rna-sgrna骨架-f、t7-pkac spacer rna-sgrna骨架-f)包含t7转录序列、spacer rna序列以及sgrna骨架首端22个碱基序列,反向引物为sgrna骨架尾端17个碱基序列(sgrna骨架-r)。
[0062]
t7-spacer rna-sgrna骨架序列通过pcr反应获得,pcr反应体系为:primestar max dna polymerase 25μl、引物f(20μm)1μl、引物r(20μm)1μl、模板1μl、ddh2o 22μl,总体积共50μl。反应条件为98℃预变性10min;98℃ 30s,58℃ 30s,72℃ 7s进行35个循环;72℃延伸7min。
[0063]
pcr产物通过琼脂糖凝胶进行分析,并通过pcr反应产物纯化试剂盒进行产物回收。回收完的序列通过t7转录试剂盒进行转录,转录方法前面已经具体说明,t7转录sgrna序列pcr引物序列如下所示。
[0064]
t7-mpka spacer rna-sgrna骨架-f:
[0065]
taatacgactcactataggggcagggtcgcaagatcttcagttttagagctagaaatagcaa;
[0066]
t7-stec spacer rna-sgrna骨架-f:
[0067]
taatacgactcactatagggtgggttgcagacacacccaagttttagagctagaaatagcaa;
[0068]
t7-pkac spacer rna-sgrna骨架-f:
[0069]
taatacgactcactatagggggagcagaaaaatcctccgagttttagagctagaaatagcaa;
[0070]
sgrna骨架-r:
[0071]
gcaccgactcggtgcca。
[0072]
t7转录完成的sgrna采用异丙醇沉淀的方法纯化收集,异丙醇沉淀体系为产物30μl、异丙醇300μl、h2o 120μl,总体积共450μl,沉淀条件为-20℃,2h。然后,13000g 4℃离心15min。去掉上清液,500μl 75%冰乙醇洗涤2次,室温吹干15~20min,最后用50μl rnase free h2o溶解,即获得sgrna。
[0073]
本发明的基因切割的片段总长设为1020bp,被切割的片段的5端位于20nt spacer rna位点前300bp,被切割的片段的3端位于20nt spacer rna位点后700bp,保证crispr-cas9切割的两个片段大小有差异,可以通过核酸凝胶电泳区分条带。
[0074]
体外切割体系为cas9酶1μl、buffer 2μl、dna模板5μl、sgrna 2μl、h2o 10μl,总体积共20μl,切割条件为37℃,6h或者过夜。sgrna体外切割后的结果如图3a所示,实验结果证明,设计的spacer rna可以有效切割靶点。
[0075]
(5)rnp的体外组装。将步骤(4)中得到的具备切割功能的sgrna与cas9酶在缓冲液中进行体外组装并于pcr仪中孵育。rnp组装体系为cas9酶6μl、buffer 4.4μl、sgrna 10μl、h2o 23.6μl、总体积共44μl,孵育条件为37℃下孵育15min~30min,得到rnp基因编辑体系。
[0076]
其中可以编辑mpka基因的体系命名为rnp-mpka,可以编辑stec基因的体系命名为rnp-stec,可以编辑pkac基因的体系命名为rnp-pkac。rnp组装完成后可以直接用于丝状真菌的转化,转化当天组装rnp。
[0077]
(6)修复片段的扩增。筛选标签(pyrg)以及rnp靶向作用的spacer rna基因位点的上下游短修复片段39bp作为同源臂,通过pcr扩增获得,并采用乙醇沉淀法收集扩增获得的片段。anpyrg的基因在ncbi数据库中的序列编号为bn001301.1,其序列通过生物公司合成,作为修复模板的pcr扩增模板。修复片段pcr扩增的正向引物(mpka修复片段-f、stec修复片段-f、pkac修复片段-f)为rnp靶向作用的spacer rna基因位点的上游39bp同源臂序列,反向引物(mpka修复片段-r、stec修复片段-r、pkac修复片段-r)为rnp靶向作用的spacer rna基因位点的下游39bp同源臂序列,引物序列由生物公司合成。
[0078]
修复模板参考步骤(4)的体系进行pcr反应获得,扩增所需的模板为生物公司合成的含有上述anpyrg基因的载体,反应条件为98℃预变性10min;98℃30s,58℃30s,72℃7s进行35个循环;72℃延伸7min。pcr产物通过琼脂糖凝胶进行分析,并通过pcr反应产物纯化试剂盒进行产物回收,修复片段pcr引物序列如下所示,其中大写字母部分为同源臂序列。
[0079]
mpka修复片段-f:
[0080]
tgttttcttctttgtacgagtgtttatcatggccgacttgcaacttcctcgagaacgcgc;
[0081]
mpka修复片段-r:
[0082]
tgtagcgctcatcgacgataaagtcctggttgaagacctcccttttagtcaataccgttaca;
[0083]
stec修复片段-f:
[0084]
tatctcgcagccatgcttgcaaaagcaacctacaacccggcaacttcctcgagaacgcgc;
[0085]
stec修复片段-r:
[0086]
ttctgcgaggacccagtgtagtaggaagttgtaggggtccccttttagtcaataccgttaca;
[0087]
pkac修复片段-f:
[0088]
cctcaagattccgtgcctcaacagtccaatcggtcttcggcaacttcctcgagaacgcgc;
[0089]
pkac修复片段-r:
[0090]
gcttgcgtcacagcggattgcatggaggctacctgaccgcccttttagtcaataccgttaca。
[0091]
乙醇沉淀法收集dna片段:500μl的pcr反应产物加入50μl的乙酸钠溶液(3m,ph5.2)以及1.25ml的无水乙醇,混匀,-20℃静置2~4h或者过夜。然后12000g,4℃离心10min,去掉上清液,留下沉淀。1ml70%的乙醇洗涤沉淀2次,每次12000g,4℃离心10min后去掉上清液。乙醇洗涤结束后置于50℃烘箱烘干。最后加入30~60μl的无菌水重悬产物,混匀后得到修复片段用于后续的实验。
[0092]
修复片段的核苷酸序列如下所示,修复片段是将spacer rna位点的前39bp上游同源序列、筛选标签和spacer rna位点的后39bp下游同源序列按顺序组装得到的,其中大写字母且划线的序列为20nt spacer rna位点的前39bp上游同源序列,小写字母的序列为anpyrg筛选标签的序列,大写字母且没有划线的序列为20nt spacer rna位点的后39bp下游同源序列。
[0093]
敲除mpka基因的修复序列如seq id no.14所示,敲除stec基因的修复序列如seq id no.15所示,敲除pkac基因的修复序列如seq id no.16所示。
[0094]
本实施例构建的rnp基因编辑体系工作原理示意图如图1所示,获得的rnp基因编辑体系体外切割作用如图3b所示,实验结果证明rnp基因编辑体系可以有效切割目的片段。因此,本实施例的rnp可以在黑曲霉中作用,能够很好的编辑黑曲霉的基因,该方法免去构建重组载体的繁琐程序,操作简单,可以高效用于黑曲霉的基因编辑。
[0095]
实施例2
[0096]
涉及的培养基以及溶液配方:
[0097]
pda培养基:300g/l马铃薯浸出粉,20g/l葡萄糖,0.1g/l氯霉素,2g/l琼脂;cd培养基:20g/l无水葡萄糖,2g/l氯化钾,1g/l磷酸二氢钾,3g/l硝酸钠,0.5g/l七水硫酸,0.01g/l七水硫酸亚铁。
[0098]
高渗cd培养基:342.3g/l蔗糖,2g/l氯化钾,1g/l磷酸二氢钾,3g/l硝酸钠,0.5g/l七水硫酸,0.01g/l七水硫酸亚铁。
[0099]
普通cd培养基:20g/l无水葡糖糖,2g/l氯化钾,1g/l磷酸二氢钾,3g/l硝酸钠,0.5g/l七水硫酸,0.01g/l七水硫酸亚铁。
[0100]
dpy培养基:20g/l无水葡萄糖,5g/l酵母粉,10g/l蛋白胨,10g/l氯化钠。
[0101]
淀粉发酵培养基:50g/l玉米淀粉,30g/l玉米浆,20g/l豆粕粉。115℃灭菌20min。
[0102]
stc溶液:10mm tris-hcl,1.2m山梨醇,50mm氯化钙,ph7.5,先定容后调节ph,0.22
μl膜过滤除菌。
[0103]
peg溶液:60%(w:v)peg4000,50mm氯化钙,10mm tris-hcl,ph7.5,先定容后调节ph,高温高压灭菌。
[0104]
调控黑曲霉菌丝形态结构的方法,包括如下步骤:
[0105]
(1)黑曲霉孢子的收集。将已经敲除pyrg筛选标签的黑曲霉cbs513.88转接到pda(马铃薯葡萄糖培养基)固体培养基中,置于30℃培养箱静置培养7d。黑曲霉cbs513.88敲除pyrg筛选标签的菌株在文献“董宏智.黑曲霉crispr基因编辑技术建立与内质网磷脂调控机制的研究[d].华南理工大学.”中公开,为本实验室保藏的材料。培养结束后,加入10ml无菌水于固体培养基上,用无菌涂布棒以涂布的形式来回涂抹加入的无菌水,使固体培养基上长出的孢子溶解在无菌水中,即可获得孢子液。
[0106]
(2)黑曲霉菌丝的富集培养。将上述收集的孢子接种于dpy(酵母粉蛋白胨葡萄糖培养基)中,置于30℃,220rpm摇床中培养2d。采用布氏漏斗,抽滤法获得菌丝,并用无菌水洗涤菌丝2次,然后再使用0.8m的氯化钠洗涤2次,称重。
[0107]
(3)黑曲霉的转化。采用peg介导的原生质体的方法进行黑曲霉的转化。将抽滤好的菌丝转移到原生质体酶解液中,酶解液的体系为1%(w:v)纤维素酶,1%(w:v)蜗牛酶,溶壁酶0.5%(w:v),1
×
磷酸钠缓冲液,0.8m氯化钠。按照1.5g菌丝加入到20ml酶液中来计算,30℃的条件下120rpm酶解。1.5h通过血球计数板测量原生质体的浓度。每隔30min镜检一次,直至原生质体的浓度达到106cfu/ml时停止酶解。该过程大概需要3h左右。酶解结束后用4层milipore过滤收集原生质体,然后再用20ml的0.8m氯化钠冲洗酶解液的瓶子及milipore。4℃,900g离心原生质体10min,弃上清。加入5ml的stc缓冲液重悬原生质体,分散均匀后再加入25ml的stc缓冲液重悬,然后再4℃的条件下900g离心收集原生质体10min该步骤重复一次。用800μl的stc缓冲液重悬原生质体,得到原生质体悬液用于后续的转化。根据不同的敲除基因,将对应的实施例1步骤(5)组装的rnp基因编辑体系44μl和实施例1步骤(6)得到的修复片段100μl加入160μl原生质体中,并加入peg溶液,用去尖枪头轻轻吹打混均匀,冰上放置30min。再加入1.5ml peg溶液后,盖上离心管盖上下颠倒使其充分混均匀,室温放置25min。期间倒好下层板,即高渗cd培养基(含2%的琼脂)。最后将上述转化混合液倒入含30℃~45℃的6ml软琼脂高渗cd培养基(含0.5%琼脂)以及3ml的stc缓冲液的15ml无菌离心管中,上下颠倒混匀。最后将混合物(上层板,含0.5%的高渗cd培养基)均匀的倒在蔗糖cd高渗固体培养基上(下层板,含2%的高渗cd培养基),30℃培养。
[0108]
(4)转化子的挑选及验证。培养6d,期间上层培养基(含0.5%的高渗cd培养基)会凝固,从转化板上挑选冒出的转化子到新的普通cd培养基中(含2%的琼脂),并于30℃培养箱中静置培养4d,由于宿主已经被敲除pyrg基因,因此只有当菌株补充pyrg基因或者其他的营养物质后,才会有菌体生长,通过pyrg作为筛选标签可以获得阳性转化子。挑选菌丝提取dna,并进行pcr验证以及测序分析,获得阳性转化子。pcr验证设计的原理如图2所示。通过rnp对基因组上面基因做出切口,然后通过上/下游39bp同源臂以及1398bp的pyrg筛选标签将修复片段(39bp上游同源臂,加1398bp的pyrg筛选标签,加39bp的下游同源臂)重组到切口位点,从而破坏基因,达到敲除基因的目的。当基因被敲除成功,通过设计的引物进行pcr扩增可以多扩增1398bp的片段,从而达到鉴定的目的。pcr鉴定的引物序列如下所示,由生物公司合成。
[0109]
mpka敲除鉴定-f:ataacttcccctttacctcccc;
[0110]
mpka敲除鉴定-r:gtcaaacctaccagacaatgcc;
[0111]
stec敲除鉴定-f:agtggcgatagcccagccttta;
[0112]
stec敲除鉴定-r:cgaggacccagtgtagtaggaa;
[0113]
pkac敲除鉴定-f:agcaaacaccccctctcagc;
[0114]
pkac敲除鉴定-r:aggtatgatgggcagacggc。
[0115]
(5)对阳性转化子进行菌落形态分析,以及菌丝形态分析。
[0116]
(6)将转化子转接于淀粉发酵培养基,并置于30℃,250rpm的摇床中进行发酵培养。发酵培养5d后,每天取发酵上清液,采用bca试剂盒测定总蛋白的含量,持续培养14d。
[0117]
本实施例中的mpka基因、stec基因、pkac基因被敲除的情况如图3c所示,均被有效敲除。
[0118]
本实施例的黑曲霉菌丝形态调控的相关基因被成功敲除的突变体的菌落形态图、菌丝形态图、黑曲霉野生菌株菌落形态图以及菌丝形态图如图4所示,其中a是野生菌株wt的菌落图,b是野生菌株wt的菌丝形态图,c是丝状真菌mapk途径上的mpka基因被敲除的突变体菌落图,d是mpka基因被敲除的突变体菌丝形态图,e是丝状真菌mapk途径上的stec基因被敲除的突变体菌落图,f是stec基因被敲除的突变体菌丝形态图,g是camp-pka途径上pkac基因被敲除的突变体菌落图,h是pkac基因被敲除的突变体菌丝形态图。可以看出,相较另外三组,敲除mpka基因的黑曲霉菌落较小,生长缓慢,菌丝呈现短杆状,基本无分支。
[0119]
本实施例的黑曲霉菌丝形态调控的相关基因被成功敲除的突变体的菌落直径以及野生菌株的菌落直径如图5所示。激酶被敲除的菌株的菌落直径显著小于野生对照菌株的菌落直径,尤其是敲除mpka基因菌株的菌落直径更小。
[0120]
另外,本实施例的黑曲霉突变体、宿主、以及野生菌株的总蛋白分泌情况如图6所示。敲除mpka基因的黑曲霉的总蛋白分泌量显著高于宿主和野生菌株。
[0121]
综合以上实验结果,得出以下结论:
[0122]
(1)rnp基因编辑体系可以在黑曲霉中作用,能够很好的编辑黑曲霉的基因,该方法免去构建重组载体的繁琐程序,操作简单,可以用于黑曲霉的基因编辑。
[0123]
(2)相比较于黑曲霉野生菌株,黑曲霉mapk途径上的stec基因,以及camp-pka途径上的pkac基因,黑曲霉mapk途径上的mpka基因可以调控菌丝形态,当敲除mpka基因时,黑曲霉菌落较小,生长缓慢,菌丝呈现短杆状,基本无分支。说明黑曲霉的mpka基因对于菌丝形态的调控具有特异性。由于被敲除mpka的菌株生长缓慢,无分支,所以可以考虑使用微流控等高通量技术,这样可以保证黑曲霉在微流控的油滴体系中避免菌丝过大导致油滴破裂,导致黑曲霉无法使用微流控。同时,菌丝形态简单,可以促进黑曲霉在流式细胞仪体系中的应用。因此,这种形态可以促进黑曲霉的高通量技术的发展,补充了黑曲霉无法使用高通量技术的短板。
[0124]
(3)黑曲霉mpka基因被敲除时,其蛋白的分泌量显著高于黑曲霉野生菌株,黑曲霉mapk途径上的stec基因敲除菌株,以及camp-pka途径上的pkac基因敲除菌株。说明敲除黑曲霉的mpka基因可以促进菌体蛋白的分泌。可能是因为菌丝结构呈现简单化,减轻了菌株的生长压力以及能量方面的输出,从而使菌体在蛋白质分泌方面得到提升。
[0125]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的
限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1