一种马铃薯早疫病致病菌茄链格孢菌的可视化检测方法
1.本发明涉及生物检测技术领域,特别是一种马铃薯早疫病致病菌茄链格孢菌的可视化检测方法。
背景技术:
2.马铃薯早疫病主要是由茄链格孢菌(alternaria solani)引起的一种严重威胁马铃薯生产的病害,在世界各处的马铃薯种植地都普遍发生。针对这种致病菌,杀菌剂的效果较为显著,但长期使用杀菌剂会导致病菌耐药性的增加。所以开发出一种能够在早期迅速而灵敏地检测alternaria solani的方法是十分重要的。
3.目前对于alternaria solani的检测方法主要是实时荧光定量pcr(quantitative real-time polymerase chain reaction,qpcr)和环介导等温扩增(loop-mediated isothermal amplification,lamp)法,qpcr法对仪器、操作人员和检测场地的高要求以及对热循环的需求,使其不能被应用于现场即时检测(point-of-care testing,poct)。基于lamp的检测方法虽然解决了以上问题,但这个方法特异性低,假阳性率高。成簇规则间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,crispr)/crispr相关(crispr-associated,cas)蛋白系统是细菌和古细菌在长期演化过程中形成的一种用来对抗入侵的病毒和外源dna的适应性免疫防御系统。这些系统在识别并切割靶标dna或靶标rna以后,会激活他们的反式切割活性,这为高特异性的、高灵敏度的核酸检测方法提供了基础。金纳米颗粒(gold nanoparticles,aunps)凭借其固有的光学稳定性、高消光系数和局部表面等离子体共振等特殊光学特性,在poct中应用前景广阔。
4.因此,亟待开发一种马铃薯早疫病致病菌茄链格孢菌的可视化检测方法。
技术实现要素:
5.本发明的目的是要解决现有技术中存在的不足,提供一种马铃薯早疫病致病菌茄链格孢菌的可视化检测方法,实现对茄链格孢菌的高特异性和高灵敏度检测,然后利用aunps实现检测结果的可视化。
6.为达到上述目的,本发明是按照以下技术方案实施的:
7.一种马铃薯早疫病致病菌茄链格孢菌的可视化检测方法,包括以下步骤:
8.s1、提取茄链格孢菌中基因组dna作为lamp反应的模板即茄链格孢菌模板dna,并进行lamp扩增;
9.s2、利用crrna特异性靶向并结合茄链格孢菌模板dna的扩增产物,激活cas12a对附近reporter dna或linker ssdna的非特异性切割活性,利用crispr/cas12a进行酶切反应,并将含有reporter dna的反应通过pcr仪实时监测荧光,保留含有linker ssdna的反应产物用于后续可视化检测;
10.s3、使用盐老化法制备了两种dna功能化的aunps,两种dna功能化的aunps通过linker ssdna连接起来而发生聚集,用肉眼观看颜色变化或者用酶标仪测量混合物的吸光
度。
11.进一步地,所述步骤s1中lamp扩增具体包括:
12.按照warmlamp kit(dna&rna)的说明进行lamp扩增,扩增反应体系为25μl,其中包含warmstart lamp 1
×
master mix,1
×
引物混合物,0.5
×
sybr greenⅰ和茄链格孢菌模板dna,其中引物混合物包括0.2μm的f3、0.2μm的b3、1.6μm的fip、1.6μm的bip、0.4μm的lf和0.4μm的lb;混匀后置于pcr仪中在65℃下反应,每1.5min采集一次荧光信号。
13.进一步地,所述步骤s2中crispr/cas12a进行酶切反应具体包括:
14.将cas12a与crrna按照浓度比cas12a:crrna=1:1.25的比例在ne bufferr2.1的depc水溶液中37℃下预组装10min,然后将形成的cas12a/crrna复合物加入到另一个反应体系中,使得最终反应体系体积为25μl,其中包含500nmreporter dna或linker ssdna,1
×
nebufferr2.1,150nmcas12a,187.5nmcrrna和和茄链格孢菌模板dna的扩增产物;于43℃反应,含有reporter dna的反应通过pcr仪实时监测荧光,保留含有linker ssdna的反应产物以用于后续可视化检测。
15.进一步地,所述步骤s3具体包括:
16.s31、于室温下分别以浓度比为dna1/dna2:tcep=1:100的比例将dna1和dna2还原,再按照浓度比为1:300的比例将aunps与经预处理的巯基dna混合室温下孵育过夜,在混合物中分批缓慢加入0.2m磷酸缓冲液(含2mnacl),使nacl最终浓度为0.2m;然后用10mm磷酸缓冲液洗涤3次,离心除去多余的dna,用上述10mm磷酸缓冲液重悬沉淀,检测重悬后的aunp-dna1和aunp-dna2的浓度,于4℃保存;
17.s32、根据所测得aunp-dna1和aunp-dna2的浓度,将aunp-dna1和aunp-dna2按浓度比1:1混合均匀后加入到步骤s2中的含有linker ssdna的反应产物里面,使体系中三种成分浓度比为aunp-dna1:aunp-dna2:linker ssdna=1:1:50,然后在反应体系中加入5m的nacl溶液,使nacl最终浓度为750mm,室温孵育5min,用肉眼观看颜色变化或者用酶标仪测量混合物的吸光度。
18.优选地,所述步骤s31中,0.2m磷酸缓冲液由2m nacl、200mm na2hpo4和200mm nah2po4组成,所述0.2m磷酸缓冲液的ph为7.4;所述10mm磷酸缓冲液由10mm na2hpo4和10mmna2hpo4组成,所述10mm磷酸缓冲液的ph为7.4。
19.优选地,所述步骤s31中离心的转速为12000rpm,离心时间为20min。
20.与现有技术相比,本发明结合lamp和crispr/cas12a实现对茄链格孢菌的高特异性和高灵敏度检测,然后利用aunps实现检测结果的可视化;该方法可以检测到10-3
ng/μl的茄链格孢菌模板dna,并且用肉眼可以见到明显颜色变化。由于操作简单,可以通过酶标仪即可实现高通量检测,前景广阔,有望于应用到现场及时检测。
附图说明
21.图1为本发明实施例1中的琼脂糖凝胶电泳结果图,其中1代表dna marker、2代表无模板对照组、3代表质粒扩增产物,4代表dna marker。
22.图2为本发明实施例1中酶切反应温度优化结果图,左侧柱状图代表无模板对照组,右侧柱状图代表能激活酶切反应的dsdna对照组,从左到右反应温度依次为34℃、37℃、40℃、43℃和46℃。
23.图3为本发明实施例1中的酶切反应cas12a浓度优化结果图,左侧柱状图代表无模板对照组,右侧柱状图代表能激活酶切反应的dsdna对照组,从左至右cas12a浓度依次为50nm、75nm、100nm、125nm、150nm和175nm。
24.图4为本发明实施例2中灵敏度试验的可视化结果图,其中1代表无模板对照组,2代表质粒dna浓度为100copies/μl,3代表质粒dna浓度为101copies/μl,4代表质粒dna浓度为102copies/μl,5代表质粒dna浓度为103copies/μl,6代表质粒dna浓度为104copies/μl,7代表质粒dna浓度为105copies/μl。
25.图5为本发明实施例3中特异性试验的可视化结果图,其中1代表无模板对照组,2代表阴性对照phytophthora infestans,3代表阴性对照phytophthora erythroseptica,4代表阴性对照synchytrium endobioticum,5代表阳性样本alternariasolani。
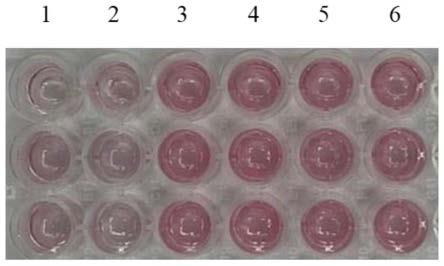
26.图6为本发明真实样本茄链格孢菌的可视化检测结果图,其中1代表无模板对照组,2代表浓度为10-4
ng/μl的菌基因组dna组,3代表浓度为10-3
ng/μl的菌基因组dna组,4代表代表浓度为10-2
ng/μl的菌基因组dna组,5代表浓度为10-1
ng/μl的菌基因组dna组,6代表浓度为100ng/μl的菌基因组dna组。
具体实施方式
27.为使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步的详细说明。此处所描述的具体实施例仅用于解释本发明,并不用于限定发明。
28.以下实施例中所用茄链格孢菌来源于北纳生物(bncc,编号337910),其他原料或试剂等未限定的均为市面上可购买,以下实施例不再描述其来源。
29.实施例1
30.1、含茄链格孢菌目标基因序列的质粒的lamp反应
31.(1)25μl lamp反应体系
32.取8μl引物fip(100μm)和bip(100μm),2μl引物lf(100μm)和lb(100μm),1μl引物f3(100μm)和b3(100μm)以及28μl depc水预混合在一起配制成10
×
引物混合物,然后取2.5μl加入到八连管中,再向其中加入0.5μl sybr greenⅰ、12.5μl warmstart lamp 2
×
master mix和1μl含有茄链格孢菌目标基因序列的的质粒dna(无模板对照组加1μl depc水),最后加入depc水将总体积定为25μl,混匀后在pcr仪中于65℃下反应,每1.5min采集一次荧光信号,最终得茄链格孢菌质粒的扩增产物。
33.(2)实验结果
34.取1μl lamp扩增产物、1μl loading buffer和4μl depc水混合均匀,在2%琼脂糖凝胶上电泳,90v电泳40min后,利用凝胶成像系统观察结果并成像。如图1所示,泳道从左至右依次代表的是dna marker、无模板对照组的扩增产物、质粒扩增产物和dna marker,从图中可以看出,质粒扩增产物所在泳道出现很多梯形条带,证明质粒成功被扩增。
35.2、crispr/cas12a酶切反应条件的优化
36.利用crrna特异性靶向并结合茄链格孢菌模板dna的扩增产物,激活cas12a对附近reporter dna的非特异性切割活性,利用crispr/cas12a进行酶切反应,并通过pcr仪实时监测荧光信号:
37.将cas12a与crrna按照浓度比cas12a:crrna=1:1.25的比例在ne buffer r2.1中
37℃下预组装10min,然后将形成的cas12a/crrna复合物加入到另一个体系中,使得最终反应体系为25μl,其中包含500nm reporter dna和含茄链格孢菌目标基因序列的质粒模板dna的扩增产物。首先对反应温度进行优化,将反应温度分别设置为34℃、37℃、40℃、43℃和46℃,得到最佳反应温度后,再对cas12a浓度进行优化,cas12a浓度分别设置为50nm、75nm、100nm、125nm、150nm和175nm。以上反应通过pcr仪实时监测荧光。
38.(1)酶切反应温度的优化
39.根据不同组酶切反应速率的快慢确定最佳反应温度,如图2所示,反应时间相同,当反应温度为43℃时,反应达到的荧光值最高,因此确定43℃为最佳酶切反应温度。
40.(2)酶切反应cas12a浓度优化
41.根据不同组酶切反应速率确定最佳cas12a浓度,如图3所示,随着cas12a浓度的增加,反应越来越快,但考虑到成本问题,确定用150nm作为cas12a最终参与反应的浓度。
42.以下实施例的crispr/cas12a酶切反应均采用实施例1确定的43℃作为最佳酶切反应温度,150nm作为cas12a最终参与反应的浓度。
43.实施例2
44.在上述实施例1的基础上,对马铃薯早疫病致病菌茄链格孢菌的可视化检测方法的灵敏度进行检测,具体如下:
45.1、25μl lamp反应体系
46.取8μl引物fip(100μm)和bip(100μm),2μl引物lf(100μm)和lb(100μm),1μl引物f3(100μm)和b3(100μm)以及28μl depc水预混合在一起配制成10
×
引物混合物,然后取2.5μl加入到八连管中,再向其中加入0.5μl sybr greenⅰ、12.5μl warmstart lamp 2
×
master mix和1μl含有茄链格孢菌目标基因序列的不同浓度的质粒dna(无模板对照组加1μl depc水),最后加入depc水将总体积定为25μl,混匀后在pcr仪中于65℃下反应,每1.5min采集一次荧光信号,进行灵敏度试验。
47.2、25μl酶切反应体系
48.取112.5μl cas12a(1μm)、140.6μl crrna(1μm)、75μl ne buffer r2.1(10
×
)和271.9μl depc水混合在一起,于37℃水浴下预组装10min,然后取20μl结合后的cas12a/crrna复合物加入八连管的孔中,再向其中分别加入2.5μl不同浓度质粒的扩增产物,最后向其中加入2.5μl linker ssdna(5μm),于43℃反应。保留反应产物用于后续可视化检测。
49.3、盐的老化
50.于室温下分别以dna1/dna2:tcep=1:100的比例将dna1和dna2还原,再按照1:300的比例将aunps与经预处理的巯基dna混合室温下孵育过夜,在混合物中分批缓慢加入0.2m磷酸缓冲液(2m nacl,200mm na2hpo4和200mm nah2po4,ph 7.4),使最终nacl浓度为0.2m。然后用10mm磷酸缓冲液(10mm na2hpo4和10mm nah2po4,ph 7.4)洗涤3次,离心(12000rpm,20min)除去多余的dna,用上述10mm磷酸缓冲液重悬沉淀,得到aunp-dna1和aunp-dna2溶液,测两个溶液浓度后于4℃保存。
51.4、可视化检测
52.根据所测得aunp-dna1和aunp-dna2的浓度,将aunp-dna1和aunp-dna2按浓度比1:1混合均匀后加入到含有linker ssdna的cas12a切割产物中,最终体系中三种成分浓度比为aunp-dna1:aunp-dna2:linker ssdna=1:1:50,然后在反应体系中加入5m nacl溶液,使
nacl最终浓度为750mm左右,室温孵育5min,用肉眼观看颜色变化或者用酶标仪测量混合物的吸光度。
53.5、实验结果
54.质粒灵敏度试验的可视化检测结果
55.如图4所示,当质粒浓度高于100copies/μl(含100copies/μl)时,通过肉眼观察,体系颜色与无模板对照组、1copies/μl和10copies/μl具有明显差异,表明反应检测限为100copies/μl。
56.实施例3
57.在上述实施例1的基础上,对马铃薯早疫病致病菌茄链格孢菌的可视化检测方法的特异性进行检测,具体如下:
58.1、25μl lamp反应体系
59.取8μl引物fip(100μm)和bip(100μm),2μl引物lf(100μm)和lb(100μm),1μl引物f3(100μm)和b3(100μm)以及28μl depc水预混合在一起配制成10
×
引物混合物,然后取2.5μl加入到八连管中,再向其中加入0.5μl sybr greenⅰ、12.5μl warmstart lamp 2
×
master mix和1μl含有茄链格孢菌目标基因序列的质粒dna(无模板对照组加入1μl depc水,阴性对照组分别加入1μl对应的阴性对照dna),混匀后在pcr仪中于65℃下反应,每1.5min采集一次荧光信号,进行特异性试验。
60.2、25μl酶切反应体系
61.取112.5μl cas12a(1μm)、140.6μl crrna(1μm)、75μl ne buffer r2.1(10
×
)和271.9μl depc水混合在一起,于37℃水浴下预组装10min,然后取20μl结合后的cas12a/crrna复合物加入八连管的孔中,再向其中分别加入2.5μl不同组的扩增产物,最后向其中加入2.5μl linker ssdna(5μm),于43℃反应。保留反应产物以用于后续可视化检测。
62.3、盐的老化
63.于室温下分别以dna1/dna2:tcep=1:100的比例将dna1和dna2还原,再按照1:300的比例将aunps与经预处理的巯基dna混合室温下孵育过夜,在混合物中分批缓慢加入2m磷酸缓冲液(2m nacl,200mm na2hpo4和200mm nah2po4,ph 7.4),使nacl最终浓度为0.2m。然后用10mm磷酸缓冲液(10mm na2hpo4和10mm nah2po4,ph 7.4)洗涤3次,离心(12000rpm,20min)除去多余的dna,用上述10mm磷酸缓冲液重悬沉淀,得到aunp-dna1和aunp-dna2溶液,测两个溶液浓度后于4℃保存。
64.4、可视化检测
65.根据所测得aunp-dna1和aunp-dna2的浓度,将aunp-dna1和aunp-dna2按浓度比1:1混合均匀后加入到含有linker ssdna的cas12a切割产物中,最终体系中三种成分浓度比为aunp-dna1:aunp-dna2:linker ssdna=1:1:50,然后在反应体系中加入5m nacl溶液,使nacl最终浓度为750mm左右,室温孵育5min,用肉眼观看颜色变化或者用酶标仪测量混合物的吸光度。
66.5、实验结果
67.①
特异性试验的可视化检测结果
68.如图5所示,从左到右依次为空白对照组、阴性对照phytophthora infestans、阴性对照phytophthora erythroseptica、阴性对照synchytrium endobioticum和阳性样本
茄链格孢菌,阳性样本与空白和三种阴性对照皆具有明显颜色差异,证明该方法对茄链格孢菌的检测具有较高特异性。
69.应用实例
70.使用本发明的方法对马铃薯早疫病致病菌茄链格孢菌真实样本进行检测,具体步骤如下:
71.1.马铃薯早疫病菌菌体的收集及基因组dna的提取
72.将茄链格孢菌置于综合pda培养基中培养3天,然后收集其菌丝,采用商业真菌dna提取试剂盒提取茄链格孢菌的基因组dna。
73.2.25μl lamp反应体系
74.取8μl引物fip(100μm)和bip(100μm),2μl引物lf(100μm)和lb(100μm),1μl引物f3(100μm)和b3(100μm)以及28μl depc水预混合在一起配制成10
×
引物混合物,然后取2.5μl加入到八连管中,再向其中加入0.5μl sybr greenⅰ、12.5μl warmstart lamp 2
×
master mix和1μl从茄链格孢菌中提取的不同浓度的dna(无模板对照组加入1μl depc水)最后加入depc水将总体积定为25μl,混匀后在pcr仪中于65℃下反应,每1.5min采集一次荧光信号,最终得真菌茄链格孢菌dna的扩增产物。
75.3.25μl酶切反应体系
76.取75μl cas12a(1μm)、93.8μl crrna(1μm)、50μl ne buffer r2.1(10
×
)和181.2μl depc水混合在一起,于37℃水浴下预组装10min,然后取20μl结合后的cas12a/crrna复合物加入八连管的孔中,再向其中加入2.5μl不同浓度真菌茄链格孢菌dna的扩增产物,最后向其中加入2.5μl linker ssdna(5μm),于43℃反应。保留反应产物以用于后续可视化检测。
77.4.盐的老化
78.于室温下分别以dna1/dna2:tcep=1:100的比例将dna1和dna2还原,再按照1:300的比例将aunps与经预处理的巯基dna混合室温下孵育过夜,在混合物中分批缓慢加入0.2m磷酸缓冲液(2m nacl,200mm na2hpo4和200mm nah2po4,ph 7.4),使nacl最终浓度为0.2m。然后用10mm磷酸缓冲液(10mm na2hpo4和10mm nah2po4,ph 7.4)洗涤3次,离心(12000rpm,20min)除去多余的dna,用上述10mm磷酸缓冲液重悬沉淀,得到aunp-dna1和aunp-dna2溶液,测两个溶液浓度后于4℃保存。
79.5.可视化检测
80.根据所测得aunp-dna1和aunp-dna2的浓度,将aunp-dna1和aunp-dna2按浓度比1:1混合均匀后加入到含有linker的cas12a切割产物中,最终体系中三种成分浓度比为aunp-dna1:aunp-dna2:linker ssdna=1:1:50,然后在反应体系中加入5m nacl溶液,使nacl最终浓度为750mm左右,室温孵育5min,用肉眼观看颜色变化或者用酶标仪测量混合物的吸光度。如图6所示,当茄链格孢菌的基因组dna浓度达到10-3
ng/μl以后,通过肉眼观察,体系的颜色与无模板对照组和10-4
ng/μl基因组dna组具有明显差异,说明本发明方法对茄链格孢菌的基因组dna的检测限为10-3
ng/μl。本发明的技术方案不限于上述具体实施例的限制,凡是根据本发明的技术方案做出的技术变形,均落入本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1