镍催化合成1,1-二取代联烯的方法
1.本发明涉及镍催化合成技术领域,具体而言,涉及一种镍催化合成1,1-二取代联烯的方法。
背景技术:
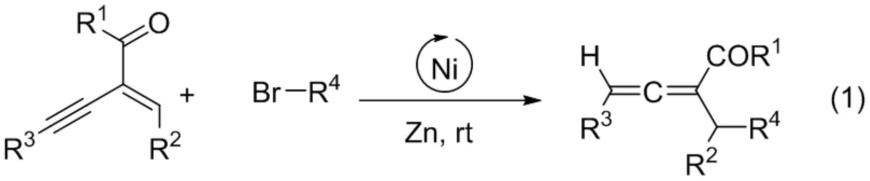
2.联烯不但是药物分子中的一类常见结构片段,也是有机合成反应中的重要官能团。联烯的合成与转化及在药物开发中的应用,已经成为有机合成和药物化学重要的研究分支。发展简单高效的催化体系,精准且模块化的合成联烯仍旧是联烯化学的重要发展方向。最近,随着自由基化学的不断发展,基于1,3-烯炔和自由基反应合成联烯已经成为重要的策略。例如,在1,3-烯炔酮为自由基受体,烷基溴代物为自由基前体,锌粉为还原剂的室温条件下,可实现镍催化的三取代联烯的合成(方程式1)。除了镍催化体系外,光催化的类似反应也有不少报导。
[0003][0004]
当反应采用双催化体系时,可实现1,3-烯炔、自由基、亲核试剂的三组分反应,高效合成四取代联烯。例如,在光和铜双催化条件下,1,3-烯炔、烷基自由基和三甲基硅氰通过三组分反应,可以高效合成氰基取代的四取代联烯(方程式2)。利用类似策略,当选用和铜催化体系相匹配的手性配体时,可以立体选择性地合成联烯。
[0005][0006]
尽管1,3-烯炔和烷基自由基的反应已成为一类合成联烯的重要方法,但该策略主要应用于三取代和四取代联烯的合成,还没有应用于1,1-二取代联烯的合成。
技术实现要素:
[0007]
本发明要解决的技术问题是提供一种镍催化合成1,1-二取代联烯的方法,以补充该领域的技术空缺,且该方法具有试剂易得、底物普适性好、官能团兼容性强、操作简便,以及反应条件温和的优点。
[0008]
为解决上述问题,本发明提供一种镍催化合成1,1-二取代联烯的方法,包括以下步骤:
[0009]
s1:往具有搅拌子的反应管中加入2-(1-炔基)-2-烯烃-1-酮、n-(酰氧基)邻苯二甲酰亚胺、镍盐、锌粉,抽真空,充入氮气保护后,进一步注入二甲亚砜;
[0010]
s2:将所述步骤s1反应后的反应管在室温下搅拌24h后结束反应,加入h2o和饱和nh4cl溶液,使反应淬灭,用乙酸乙酯萃取混合物,再用饱和食盐水反萃取一次有机相;
[0011]
s3:收集所述步骤s2得到的有机层,用无水硫酸镁干燥,进一步过滤后,得到有机相溶液,并通过旋转蒸发仪除去其溶剂;
[0012]
s4:用石油醚和乙酸乙酯的混合液作为淋洗剂,对所述步骤s3处理后的所述有机相溶液进行柱层析分离,最后经旋转蒸发仪蒸馏得到1,1-二取代联烯。
[0013]
作为优选的方案,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮与镍盐的摩尔比为1︰0.1-0.2。
[0014]
作为优选的方案,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮与锌粉的摩尔比为1︰2-3。
[0015]
作为优选的方案,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮与n-(酰氧基)邻苯二甲酰亚胺的摩尔比为1︰1.5-2.0。
[0016]
作为优选的方案,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮与二甲亚砜的摩尔比为1︰200-300。
[0017]
作为优选的方案,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮为1-三甲基硅乙炔基-1-对甲氧基苯甲酰基-2-苯基乙烯。
[0018]
作为优选的方案,所述步骤s1中,所述镍盐为醋酸镍四水合物。
[0019]
作为优选的方案,所述步骤s4中,所述石油醚和乙酸乙酯的混合液中石油醚和乙酸乙酯的体积比为50︰1。
[0020]
作为优选的方案,,所述步骤s4中,所述柱层析分离中硅胶柱的长度为10cm。
[0021]
作为优选的方案,所述的镍催化合成1,1-二取代联烯的反应通式如下:
[0022][0023]
本发明的有益效果是:本发明一种镍催化合成1,1-二取代联烯的方法首次发展了用镍催化的自由基加成—负离子环合反应策略,高效合成了1,1-二取代联烯;本发明一种镍催化合成1,1-二取代联烯的方法具有试剂易得、底物普适性好、官能团兼容性强、操作简便,以及反应条件温和的优点。
具体实施方式
[0024]
下面将对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0025]
本发明提供了一种镍催化合成1,1-二取代联烯的方法,包括以下步骤:
[0026]
s1:往具有搅拌子的反应管中加入2-(1-炔基)-2-烯烃-1-酮、n-(酰氧基)邻苯二甲酰亚胺、镍盐、锌粉,抽真空,充入氮气保护后,进一步注入二甲亚砜;
[0027]
s2:将所述步骤s1反应后的反应管在室温下搅拌24h后结束反应,加入h2o和饱和nh4cl溶液,使反应淬灭,用乙酸乙酯萃取混合物,再用饱和食盐水反萃取一次有机相;
[0028]
s3:收集所述步骤s2得到的有机层,用无水硫酸镁干燥,进一步过滤后,得到有机相溶液,并通过旋转蒸发仪除去其溶剂;
[0029]
s4:用石油醚和乙酸乙酯的混合液作为淋洗剂,对所述步骤s3处理后的所述有机相溶液进行柱层析分离,最后经旋转蒸发仪蒸馏得到1,1-二取代联烯。
[0030]
优选的,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮与镍盐的摩尔比为1︰0.1-0.2。
[0031]
优选的,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮与锌粉的摩尔比为1︰2-3。
[0032]
优选的,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮与n-(酰氧基)邻苯二甲酰亚胺的摩尔比为1︰1.5-2.0。
[0033]
优选的,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮与二甲亚砜的摩尔比为1︰200-300。
[0034]
优选的,所述步骤s1中,所述2-(1-炔基)-2-烯烃-1-酮为1-三甲基硅乙炔基-1-对甲氧基苯甲酰基-2-苯基乙烯。
[0035]
优选的,所述步骤s1中,所述镍盐为醋酸镍四水合物。
[0036]
优选的,所述步骤s4中,所述石油醚和乙酸乙酯的混合液中石油醚和乙酸乙酯的体积比为50︰1。
[0037]
优选的,所述步骤s4中,所述柱层析分离中硅胶柱的长度为10cm。
[0038]
优选的,所述的镍催化合成1,1-二取代联烯的反应通式如下:
[0039][0040]
下面用将提供结合具体数据以及具体材料种类的具体实施例对本发明做进一步详细说明,但本发明包括但不限于以下具体实施例:
[0041]
实施例一
[0042][0043]
s1:将1-三甲基硅乙炔基-1-对甲氧基苯甲酰基-2-苯基乙烯(0.2mmol,1.0当量),n-(金刚烷甲酰氧基)邻苯二甲酰亚胺(0.4mmol,2.0当量),ni(oac)2·
4h2o(20mol%),zn(0.4mmol,2.0当量)加入到干燥的反应管中,再加入dmso(4ml)。抽真空并回充氮气三次;
[0044]
s2:将反应混合物在室温下搅拌24h,加入3ml h2o和3ml饱和nh4cl溶液,使反应淬
灭,用乙酸乙酯萃取混合物(3
×
2ml),再用饱和食盐水反萃取一次有机相;
[0045]
s3:收集有机层,用无水硫酸钠干燥,进一步过滤后,得到有机相溶液,并通过旋转蒸发仪除去其溶剂;
[0046]
s4:经过滤浓缩后,用硅胶柱层析纯化。石油醚与乙酸乙酯的混合液(v
石油醚
:v
乙酸乙酯
=50:1)柱层析得目标产物72.5mg,收率为91%。分析数据如下:无色透明液体。1h nmr(500mhz,cdcl3)δ7.76(d,j=8.7hz,2h),7.42-7.26(m,4h),7.23-7.20(m,1h),6.86(d,j=8.8hz,2h),5.20(q,j=13.5hz,2h),3.83(s,3h),3.78(s,1h),1.95(s,3h),1.68(d,j=2.0hz,6h),1.67-1.56(m,6h).
13
c nmr(126mhz,cdcl3)δ217.6,193.6,162.8,140.5,131.7,130.4,130.3,127.4,126.3,113.0,107.3,81.2,55.3,54.2,40.4,37.5,36.9,28.7.hrms(esi)[m+h]+:calculated for c
28h31
o2:399.2324,found399.2327.。
[0047]
实施例二
[0048][0049]
s1:将1-三甲基硅乙炔基-1-对甲氧基苯甲酰基-2-苯基乙烯(0.2mmol,1.0当量),n-(甲氧基取代乙酰氧基)邻苯二甲酰亚胺(0.4mmol,2.0当量),ni(oac)2·
4h2o(20mol%),zn(0.4mmol,2.0当量)加入到干燥的反应管中,再加入dmso(4ml)。抽真空并回充氮气三次;
[0050]
s2:将反应混合物在室温下搅拌24h,加入3ml h2o和3ml饱和nh4cl溶液,使反应淬灭,用乙酸乙酯萃取混合物(3
×
2ml),再用饱和食盐水反萃取一次有机相;
[0051]
s3:收集有机层,用无水硫酸钠干燥,进一步过滤后,得到有机相溶液,并通过旋转蒸发仪除去其溶剂;
[0052]
s4:经过滤浓缩后,用硅胶柱层析纯化。用石油醚与乙酸乙酯的混合液(v
石油醚
:v
乙酸乙酯
=50:1)柱层析得目标产物43.8mg,收率为71%。分析数据如下:黄色油状液体。1h nmr(500mhz,cdcl3)δ7.79-7.76(m,2h),7.36-7.34(m,2h),7.31-7.28(m,2h),7.22-7.19(m,1h),6.87-6.84(m,2h),5.24-5.16(m,2h),4.38-4.35(m,1h),3.83(s,3h),3.78-3.75(m,1h),3.71-3.68(m,1h),3.35(s,3h).
13
c nmr(126mhz,cdcl3)δ215.5,192.0,162.9,140.6,131.5,130.7,128.4,128.1,126.8,113.1,107.8,81.0,75.1,58.7,55.3,43.3.hrms(esi)[m+h]
+
:calculated for c
20h21
o3:309.1491,found 309.1495.。
[0053]
实施例三
[0054][0055]
s1:将1-三甲基硅乙炔基-1-对甲氧基苯甲酰基-2-苯基乙烯(0.2mmol,1.0当量),n-(特戊酰氧基)邻苯二甲酰亚胺(0.4mmol,2.0当量),ni(oac)2·
4h2o(20mol%),zn(0.4mmol,2.0当量)加入到干燥的反应管中,再加入dmso(4ml)。抽真空并回充氮气三次;
[0056]
s2:将反应混合物在室温下搅拌24h,加入3ml h2o和3ml饱和nh4cl溶液,使反应淬灭,用乙酸乙酯萃取混合物(3
×
2ml),再用饱和食盐水反萃取一次有机相;
[0057]
s3:收集有机层,用无水硫酸钠干燥,进一步过滤后,得到有机相溶液,并通过旋转蒸发仪除去其溶剂;
[0058]
s4:经过滤浓缩后,用硅胶柱层析纯化。用石油醚与乙酸乙酯的混合液(v石油醚:v乙酸乙酯=50:1)柱层析得目标产物41.6mg,收率为65%。分析数据如下:白色固体。1h nmr(500mhz,cdcl3)δ7.76-7.73(m,2h),7.45-7.42(m,2h),7.29-7.26(m,2h),7.22-7.19(m,1h),6.87-6.85(m,2h),5.21(s,2h),3.95(s,1h),3.83(s,3h),1.04(s,9h).
13
c nmr(126mhz,cdcl3)δ217.3,193.5,162.8,141.5,132.3,132.2,131.7,131.5(3),131.5(0),130.4,130.1,128.5,128.4,127.5,126.3,113.0,108.4,81.4,55.3,53.2,35.7,28.4.hrms(esi)[m+h]
+
:calculated for c
22h25
o2:321.1855,found321.1850.。
[0059]
通过上述实施例,也是进一步地证明了,本发明提供了提供一种普适性好、官能团兼容性强、反应条件温和且具环保高效等优点的镍催化合成1,1-二取代联烯的方法。
[0060]
以上仅是本发明的特征实施范例,对本发明保护范围不构成任何限制。凡采用同等交换或者等效替换而形成的技术方案,均落在本发明权利保护范围之内。
[0061]
虽然本公开披露如上,但本公开的保护范围并非仅限于此。本领域技术人员,在不脱离本公开的精神和范围的前提下,可进行各种变更与修改,这些变更与修改均将落入本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1