二茂铁衍生物及其制备方法和用途与流程
二茂铁衍生物及其制备方法和用途
1.本技术是申请日为2021年2月24日,申请号为202110206235.6,发明名称为“二茂铁衍生物及其制备方法和用途”的发明专利申请的分案申请。
技术领域
2.本发明属于化合物领域,具体涉及一种二茂铁衍生物及其制备方法和用途。
背景技术:
3.癌症已成为世界范围内最重要的致死性疾病。癌症可于任何年龄在各种器官及组织中发生,导致死亡的主要癌症种类有:肺癌、胃癌、肝癌、结肠癌和乳腺癌等。虽然部分小分子抗癌药物已用于临床,部分化合物正在进行临床前的研究。大部分癌症患者发觉病情时已是中期至晚期,临床治疗总体效果较差,尤其是多药耐药性的不断出现,使得癌症的治疗困难重重。因此,开发出活性高、副作用低的新型抗癌药物来满足临床的需求迫在眉睫。
4.二茂铁(ferrocene)是一种具有独特的夹心结构化合物,二价铁离子被夹在两个平面环之间互为交错构型。二茂铁及其衍生物因其自身的特点:(1)芳香性,能发生取代反应,易进行修饰;(2)亲脂性,能够通过细胞膜与细胞内的各种酶相互作用;(3)低毒性,能够在体内进行代谢。二茂铁衍生物在医学领域显示出广泛的药理活性,尤其是在抗肿瘤领域药理活性尤为突出:a.rosenefeld等研究表明二茂铁修饰的顺铂衍生物具有相当强的抑制白血病活性,而且其肾毒比cis-ddp低的多(a.rosenfeld,etal.inorg.chim.acat.1992,201:219);e.w.neuse等人研究表明二茂铁衍生物具有独特的抗肿瘤、抗癌活性(e.w.neuse.j.inorga.organoment.polymers and materials.2005,15(1):3-32);x.f.huang等合成了一系列含吡唑环的二茂铁衍生物,活性研究表明部分化合物具有比5-氟尿嘧啶强的抗癌活性(x.f.huang,etal.j.organomet.chem.2012,706-707:113-123);w.liu等合成了一系列二茂铁脲衍生物,活性研究表明部分化合物具有较强的抑制hiv-1蛋白酶活性(w.liu,et al.appl.organomet.chem.2012,26:189-193);美国专利文献8426462b2公开了含芳香环的二茂铁衍生物对人乳腺癌细胞株mda-mb-231和前列腺癌细胞株pc-3具有很强的抑制活性。
5.二茂铁作为抗肿瘤药物设计合成的先导化合物(e.w.neuse.j.inorg.organoment.p.2005,15(1):3-32;s.s.braga,et al.organometallics,2013,32:5626-5639)。异噁唑杂环是一种具有潜在生物活性的药效团,通常被引入药物分子以提高活性。在申请人前期研究中,将异噁唑杂环引入二茂铁母核,合成了一系列结构新颖的含异噁唑杂环的二茂铁衍生物,并研究了其初步的体外抑肺癌细胞株a549、大肠癌细胞株hct-116和乳腺癌细胞株mcf-7活性,结果表明大部分化合物对a549,hct116和mcf-7细胞株具有很强的抑制活性(雍建平,等.申请公开号cn103601762a)。基于前期良好的研究基础,为了丰富该类化合物的种类,申请人继续设计合成该类含异噁唑杂环的二茂铁衍生物,以期发现新的抗癌先导化合物或候选化合物。
技术实现要素:
6.本发明提供一种式(i)所示的二茂铁衍生物、或其药学上可接受的盐、或其溶剂化物:
[0007][0008]
其中:z选自nh、o或s;
[0009]
r1选自氢、c1~c6烷基、卤素;
[0010]
r2独立地选自氢、卤素、c1~c6烷基、c1~c6烷氧基、卤代c1~c6烷氧基、卤代c1~c6烷基或硝基;
[0011]
n为0-5的整数,当n大于1时,r2可以为相同或不同的基团。
[0012]
根据本发明的实施方案,r1选自氢、甲基、氯、氟。
[0013]
根据本发明的实施方案,r2独立地选自下述基团中的至少一种:氢、氟、氯、溴、甲基、乙基、甲氧基、三氟甲基、叔丁基、氰基和硝基,
[0014]
根据本发明的实施方案,n为1、2或3。
[0015]
根据本发明的实施方案,术语“c1~c6烷基”可选自碳数为1,2,3,4,5,6的烷基,其余术语(例如c1~c6烷氧基)中所具有的c1~c6烷基部分同此定义。
[0016]
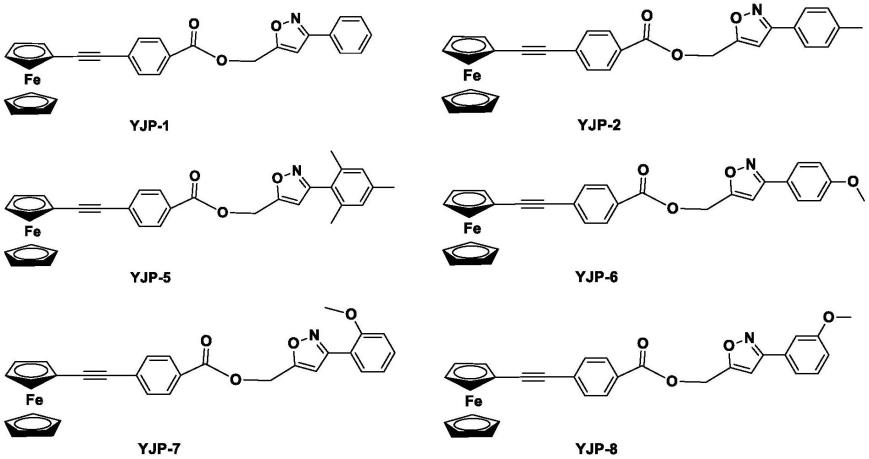
根据本发明的实施方案,所述式(i)所示的二茂铁衍生物为下述任一种化合物:
[0017]
[0018]
[0019]
[0020]
[0021]
[0022]
[0023]
[0024]
[0025]
[0026][0027]
根据本发明的实施方案,式(i)所示的二茂铁衍生物药学上可接受的盐,可以为由式(i)化合物与药学上可接受的酸或药学上可接受的阳离子形成药学上可接受的盐。其中所述药学上可接受的盐包括但不限于与无机酸形成的盐,如盐酸盐、磷酸盐、二磷酸盐、氢溴酸盐、硫酸盐、亚磺酸盐、硝酸盐、及其类似盐;也包括与有机酸形成的盐,如乳酸、草酸、苹果酸盐、马来酸盐、富马酸盐、酒石酸盐、琥珀酸盐、柠檬酸盐、乳酸盐、磺酸盐、对甲苯磺酸盐、2-羟乙基磺酸盐、苯甲酸盐、水杨酸盐、硬脂酸盐、三氟乙酸或氨基酸和链烷酸盐(如醋酸盐,hooc-(ch2)n-cooh(其中n是1-4的整数)的盐),及其类似盐。其中,所述药学上可接受的阳离子包括但不限于钠、钾、钙、铝、锂和铵。
[0028]
根据本发明的实施方案,所述溶剂化物包括水合物和醇合物。
[0029]
本发明还提供了上述式(i)所示的二茂铁衍生物的制备方法,所述的方法包括如下的步骤:
[0030]
(1)二茂铁乙炔与式a化合物反应,得到含二茂铁的中间体b;
[0031]
所述式a化合物为3-(r1)-4-溴苯甲酸,其结构式为:
[0032]
所述式b化合物的结构式为:
[0033]
其中,r1具有如上文所述的选择;
[0034]
(2)所述中间体b与式c化合物反应,得到式(i)所示的二茂铁衍生物;
[0035]
所述化合物c的结构式为:其中,r2和n具有如上文所述的选择,z’代表nh2、oh或sh;
[0036]
优选地,所述式c化合物为3-取代苯基-5-羟甲基-异噁唑(ii)、3-取代苯基-5-巯基甲基-异噁唑或3-取代苯基-5-氨甲基-异噁唑(iii)。
[0037]
根据本发明的实施方案,步骤(1)所述反应在钯(ii)化合物、有机磷和铜(i)化合物存在下进行。例如,所述钯(ii)化合物可以选自本领域已知钯(ii)化合物,比如为双三苯基磷二氯化钯、四(三苯磷)化钯和/或二苯基磷二茂铁二氯化钯;例如,所述有机磷可以选自本领域已知有机磷,比如为三苯基磷;例如,所述铜(i)化合物可以选自本领域已知铜(i)化合物,比如为碘化亚铜。
[0038]
根据本发明的实施方案,步骤(1)和步骤(2)所述的反应均在干燥的有机溶剂中进行。例如,所述干燥的有机溶剂可以选自:芳烃、卤代烃、四氢呋喃(thf)、二甲亚砜(dmso)、二氧六环、乙腈、吡啶、dmf或离子液体;优选选自四氢呋喃、氯仿、1,2-二氯甲烷、苯、甲苯、二甲苯、乙腈、吡啶或、dmf或离子液体;更优选为四氢呋喃。
[0039]
根据本发明的实施方案,步骤(1)的反应体系中还含有碱性缚酸剂。优选地,所述的碱性缚酸剂选自有机碱和/或无机碱。例如,所述有机碱选自三乙胺、三丙胺、dmap、dmf、n-甲基吗啉等中的一种、两种或更多种;例如,所述的无机碱选自碳酸钾、氢化钠、碳酸钠等中的一种、两种或更多种。更优选地,所述碱性缚酸剂为三乙胺。
[0040]
根据本发明的实施方案,步骤(1)中所述二茂铁乙炔与干燥的有机溶剂和碱性缚酸剂混合物的摩尔体积比为(0.5-5)mmol:6ml,例如0.952mmol:6ml。
[0041]
根据本发明的实施方案,步骤(1)包括如下过程:二茂铁乙炔与式a化合物分散于干燥的有机溶剂和碱性缚酸剂的混合物中,搅拌条件下向所述混合物中加入钯(ii)化合物、有机磷和铜(i)化合物,继续搅拌,然后回流反应,将反应液过滤后、浓缩滤液,得到所述中间体b。
[0042]
根据本发明的实施方案,步骤(1)和步骤(2)所述反应均在惰性气氛保护下进行,例如所述惰性气氛为氮气。
[0043]
根据本发明的实施方案,步骤(2)中所述反应在干燥的有机溶剂中进行。其中,所述有机溶剂具有如上文所示的选择。
[0044]
根据本发明的实施方案,步骤(2)中所述反应在缩合剂存在进行。例如,所述缩合剂可以选自选自dcc、dmap、nmm、hobt、hatu中的一种、两种或更多种;例如,所述缩合剂可以选自dcc和dmap的组合,dcc、hobt和dmap的组合、dcc、hobt和nmm的组合、dcc和nmm的组合或dcc和hatu的组合。
[0045]
根据本发明的实施方案,步骤(2)中所述3-取代苯基-5-羟甲基-异噁唑(ii)或3-取代苯基-5-氨甲基-异噁唑(iii)为已知化合物,可以按照公开号为cn103360382a的中国专利文献中优化的过程制备得到。具体地,制备路线如下所示:
[0046][0047]
当z’为sh时,化合物c为3-取代苯基-5-巯基甲基-异噁唑,其制备过程包括:以丙炔硫醇为原料,按照化合物(ii)的合成过程制备得到3-取代苯基-5-巯基甲基-异噁唑。
[0048]
根据本发明的实施方案,步骤(1)或步骤(2)所述反应的温度在-20℃至回流条件范围内的任意温度点,优选0℃至回流条件范围内的任意温度点,还优选室温至回流温度范围内的任意温度点。
[0049]
根据本发明的实施方案,步骤(2)包括如下过程:将所述中间体b加入干燥的有机溶剂中,而后向其中加入缩合剂反应,再向其中加入所述式c化合物反应,得到所述式(i)所示的二茂铁衍生物。优选地,加入缩合剂反应的时间为20-40min,例如30min。
[0050]
优选地,加入式c化合物反应的时间为20-40min,例如30min。
[0051]
优选地,所述式(i)化合物的合成路线如下:
[0052][0053]
如果需要,可以将式c化合物中的任何官能团予以保护;
[0054]
并且其后,如果有必要(以任何次序):
[0055]
(a)除去任何保护剂,和
[0056]
(b)形成式(i)化合物的药物组合物。
[0057]
本发明还提供一种药物组合物,其含有如式(i)所示的二茂铁衍生物,或其药学上可接受的盐、或其溶剂化物。
[0058]
根据本发明的实施方案,所述药物组合物以及含有至少一种药学上可接受的药用辅料;例如,至少一种药学上可接受的、惰性的、无毒的所述药用辅料可以选自赋形剂、载体和/或稀释剂。其中,所述药学上可接受的药用辅料是指惰性、无毒的药用辅料。
[0059]
根据本发明的实施方案,所述的药用辅料还可以选自下述辅料中的一种或多种:填充剂、崩解剂、润滑剂、助流剂、泡腾剂、矫味剂、防腐剂和包衣材料的药学可接受的辅助材料。
[0060]
本发明还提供一种药物制剂,其包含如式(i)所示的二茂铁衍生物,或其药学上可接受的盐、或其溶剂化物。
[0061]
根据本发明的实施方案,所述药物制剂含有上述药物组合物。
[0062]
根据本发明的实施方案,所述药物制剂为固体口服制剂、液体口服制剂或注射剂。
[0063]
优选地,所述的制剂选自片剂、分散片、肠溶片、咀嚼片、口崩片、胶囊、颗粒剂、口服溶液剂、注射用水针、注射用冻干粉针、大输液或小输液。
[0064]
本发明还提供一种用作药物的权利要求1-3的式(i)所示的二茂铁衍生物或其药学上可接受的盐,尤其是一种有效用于治疗肿瘤/癌症的药物或先导化合物。
[0065]
本发明还提供一种如式(i)所示的二茂铁衍生物、其药学上可接受的盐、其溶剂化物、或所述药物组合物在制备治疗抗肿瘤或抗癌药物中的应用。
[0066]
本发明还提供式(i)所示的二茂铁衍生物、其药学上可接受的盐或其溶剂化物作为抗肿瘤/癌症的先导化合物的应用。
[0067]
优选地,所述的肿瘤或癌症选自:膀胱癌、卵巢癌、乳腺癌、胃癌、食道癌、肺癌、头颈癌、结肠癌、咽癌和胰腺癌等中的至少一种;优选地,所述肺癌为非小细胞肺癌;更优选地,所述肿瘤或癌症为非小细胞肺癌、胃癌、乳腺癌和/或宫颈癌。
[0068]
本发明还提供一种预防和/或治疗上述肿瘤/癌症相关疾病的方法,包括将有效量的式(i)所示的二茂铁衍生物、其药学上可接受的盐、其溶剂化物、所述药物组合物、或所述药物制剂给予需要的患者,例如人。
[0069]
术语“有效量”指的是,所述至少一种化合物和/或至少一种药学上可接受的盐对于能有效“治疗”个体的一种疾病或不适的用量。如果是癌症时,有效量能减少癌症或肿瘤细胞的数目;缩小肿瘤的大小;抑制或阻止肿瘤细胞向周边器官的侵入,例如,肿瘤蔓延入软组织或骨骼中;抑制或阻止肿瘤的转移;抑制或阻止肿瘤的生长;一定程度上减轻一种或多种与癌症相关的症状;减少发病率和死亡率;提高生活质量;或者是上述效果的结合。有效量可以是通过抑制egfr活性来减少疾病症状的用量。对于癌症治疗,体内实验的效果可以通过评估如存活期、疾病进展时间(time to diseaseprogression,ttp)、反应率(response rates,rr)、持续反应期和/或生活质量来测量。本领域技术人员能够理解,有效量可以随着给药的途径、赋形剂的剂量、以及与其他药物的合用而变化。
[0070]
术语“有效量”还可指的是所述至少一种化合物和/或其至少一种药学上可接受的盐对抑制egfr的过度表达和/或活性过高有效的剂量。
[0071]
本发明的有益效果:
[0072]
本发明提供了如式(i)所示的结构新颖的二茂铁衍生物。所述二茂铁衍生物对肿瘤或癌症具有良好的抑制作用。根据体外抑人肺癌细胞株(a549)、乳腺癌细胞株(mcf-7)及宫颈癌细胞株(hela)研究结果表明:该类化合物对人肺癌细胞株(a549)、乳腺癌细胞株(mcf-7)及宫颈癌细胞株(hela)具有较强的抑制活性。可作为抗癌药物的候选化合物或先导化合物。
具体实施方式
[0073]
下面结合实施例对本发明作进一步的说明。需要说明的是,下述实施例不能作为对本发明保护范围的限制,任何在本发明基础上做出的改进都不违背本发明的精神。
[0074]
其中,中间体和目标化合物的合成过程均以实施例中的代表说明,其余的中间体和目标化合物的合成过程同代表化合物。
[0075]
仪器与试剂:
[0076]
avance iii核磁共振仪(400mhz,dmso-d6,tms为内标),离子阱液质连用仪(decax-30000lcq deca xp),shimadzu ftir-8400s(日本岛津制作所生产),xt5数字显示显微熔点测定仪(北京市科仪电光仪器厂制造),可调波长式微孔板酶标仪(molecular devies spectramax190).
[0077]
实施例1中间体3-取代苯基-5-羟甲基-异噁唑(ii)及3-取代苯基-5-氨甲基-异噁唑(iii)的合成
[0078]
以取代苯甲醛为原料,通过合成肟、1,3-偶极环加成反应、甲磺酰酯化反应、叠氮化、还原反应制备(r2选自氢、卤素、c1~c6烷基、c1~c6烷氧基、卤代c1~c6烷基或硝基;n为0-5的整数),具体见如下路线:
[0079][0080]
中间体3-取代苯基-5-羟甲基-异噁唑(ii)及3-取代苯基-5-氨甲基-异噁唑(iii)的具体的合成过程详见申请人在前期申请公开号为cn103360382a、cn103664991a和cn103601762a。
[0081]
实施例2含二茂铁环的中间体b的合成过程(以二茂铁乙炔和对溴苯甲酸的合成为示例):
[0082][0083]
将2.00g(9.52mmol)二茂铁乙炔和1.91g(9.52mmol)4-溴苯甲酸加入250ml双口圆底烧瓶中,接着加入干燥的四氢呋喃和三乙胺60ml,该反应物在氮气保护下,室温搅拌10分钟,接着将0.2g(0.76mmol)三苯基磷,0.28g(0.38mmol)双三苯基磷二氯化钯和0.07g(0.38mmol)碘化亚铜加入反应体系,反应体系在室温下搅拌20分钟,然后回流反应,整个反应在氮气保护下进行,tlc检测反应完成后,反应混合物过滤,滤液浓缩即得粗产品,粗产品柱分离(v
石油醚
:v
乙酸乙酯
=5:1~1:1)即得纯品4-二茂铁乙炔基-苯甲酸,2.53克,产率:81%,深浅黄色固体。4-二茂铁乙炔基-苯甲酸的1h nmr(400mhz,dmso-d6):4.29(s,5h,η
5-c5h5),4.38(2h,t,j=2.0hz),4.61(2h,t,j=2.0hz),7.58(2h,d,j=9.2hz),7.90(2h,d,j=9.2hz),12.83(1h,brs,-cooh)。
[0084]
改变r1,3-(r1)-4-溴苯甲酸制备其他中间体的制备过程参照二茂铁乙炔和4-溴苯甲酸的反应过程。
[0085]
实施例3式(i)所示的酯类目标化合物(yjp-1)的合成过程
[0086][0087]
将0.165g(0.5mmol)实施例2制备的4-二茂铁乙炔基-苯甲酸和8ml干燥的thf加入50ml单口圆底烧瓶,搅拌下将0.103g(0.5mmol)dcc和0.061g(0.5mmol)dmap加入反应体系,0℃反应30min后,接着将0.088g(0.5mmol)3-苯基-5-羟甲基-异噁唑加入反应体系,0℃反应30min后自然升至室温反应,整个反应过程在氮气保护下进行。tlc检测反应完成后,反应液减压浓缩,残渣柱分离v
(石油醚)
:v
(乙酸乙酯)
=5:1~2:1)即得目标化合物(yjp-1)。
[0088]
其余的化合物yjp-2到yjp-76参照目标化合物yjp-1的合成过程合成。
[0089]
实施例4式(i)所示的酰胺类目标化合物(yjp-77)的合成过程
[0090][0091]
将0.165g(0.5mmol)实施例2制备的4-二茂铁基-苯甲酸和8ml干燥的thf加入50ml单口圆底烧瓶,搅拌下将0.103g(0.5mmol)dcc,0.068g(0.5mmol)hobt和0.061g(0.5mmol)dmap加入反应体系,0℃反应30min后,接着将0.087g(0.5mmol)3-苯基-5-氨甲基-异噁唑加入反应体系,0℃反应30min后自然升至室温反应,整个反应过程在氮气保护下进行。tlc检测反应完成后,反应液减压浓缩,残渣柱分离v
(石油醚)
:v
(乙酸乙酯)
=5:1~2:1)即得目标化合物(yjp-77)。
[0092]
其余的yjp-78到yjp-152化合物参照目标化合物yjp-77的合成过程合成。
[0093]
[0094]
[0095]
[0096]
[0097]
[0098]
[0099]
[0100]
[0101]
[0102]
[0103][0104]
化合物yjp-1至yjp-152的结构均通过1h nmr分析方法进行了表征。yjp-1至yjp-152化合物的编号及核磁表征结果如表1所示:
[0105][0106]
表1.式i所示化合物的1h nmr
[0107]
[0108]
[0109]
[0110]
[0111]
[0112]
[0113]
[0114]
[0115]
[0116]
[0117]
[0118]
[0119]
[0120]
[0121]
[0122]
[0123]
[0124]
[0125][0126]
实施例5体外抗肿瘤活性测试
[0127]
采用cck-8法对如上实施例中的化合物进行了体外抗肿瘤活性测试。主要研究了其对乳腺癌细胞株(mcf-7)、肺腺癌细胞株(a549)和宫颈癌细胞株(hela)抑制活性的体外抑制活性。乳腺癌细胞株(mcf-7)、肺腺癌细胞株(a549)和宫颈癌细胞株(hela)来自于宁夏医科大学保存的细胞系。具体的测试过程以乳腺癌mcf-7细胞株的测试过程为例进行阐述:
[0128]
(1)乳腺癌细胞株(mcf-7)的培养及抑制活性测试过程
[0129]
将乳腺癌细胞株mcf-7置于37℃,饱和湿度,含有5%的co2培养箱中培养24小时,当细胞处于对数生长期时,吸弃上层培养液,并用0.25%胰蛋白酶-edta溶液消化后,使用高糖培养基终止消化。并将细胞接种于96孔板中,使得细胞密度为5000个/孔。将96孔板置于培养箱中培养24小时。随之吸弃96孔板中的细胞培养液。并向96孔板中补加100μl的高糖培养基,然后每孔加入不同浓度的测试样品1μl(每个浓度设置5个复孔),接着置于37℃,饱和湿度,5% co2的培养箱中继续培养48h后,每孔加入10μl cck8,继续在37℃培养箱中孵育1-4h后。在多功能酶标仪上测定450nm波长下每孔的吸光度值。按照抑制率%=[(od
对照细胞-od
加药细胞
)/(od
对照细胞-od
空白
)]
×
100。阴性对照为v
高糖培养基
/v
dmso
:10:1的混合溶液。
[0130]
(2)肺癌细胞株(a549)和宫颈癌细胞株(hela)的培养及抑制活性测试过程
[0131]
抑制肺癌细胞株a549和宫颈癌细胞株(hela)的实验过程同乳腺癌细胞株(mcf-7)的筛选过程。
[0132]
优选的化合物乳腺癌细胞株mcf-7、人肺癌细胞株a549、宫颈癌细胞株hela的活性结果分别见下表2、表3、表4。
[0133]
表2.式(i)中部分实施例化合物抑制乳腺癌细胞株mcf-7活性测试结果
[0134]
[0135][0136]
表3.式(i)中部分实施例化合物抑制人肺癌细胞株a549活性测试结果
[0137]
化合物编号浓度(μm)抑制率(%)yjp-1416.2257.00yjp-1712.4360.42yjp-1811.3579.29
[0138]
表4.式(i)中部分实施例化合物抑制宫颈癌细胞株hela活性测试结果
[0139]
化合物编号浓度(μm)抑制率(%)yjp-410.6860.10yjp-139.9174.80yjp-1416.2268.26yjp-1712.4357.62yjp-1811.3563.99yjp-1910.5388.94
[0140]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1