一种光催化的恩/达格列净前体及其合成方法与流程
1.本发明属于药物化学领域,涉及一种光催化的恩/达格列净的中间体,同时,还涉及该中间体的合成方法。
技术背景
2.碳-芳基糖苷类sglt-2抑制剂是一类新型抗糖尿病药物,其作用机制为通过抑制肾脏对葡萄糖的重吸收,使得过量的葡萄糖从尿液中排除,直接降血糖。这类药物不仅能够起到控制血糖水平的作用,在最新的研究中发现,sglt-2抑制剂适应于成人心衰患者,降低心血管死亡和心衰恶化的风险,并改善心衰症状,目前恩/达格列净均已经获得美国fda的批准降低成人hfref患者的心血管死亡和心衰住院风险,无论患者伴或不伴糖尿病。
3.目前报道的碳-芳基糖苷类sglt-2抑制剂的合成方法主要有两类:1.以溴/碘代芳烃为原料,制备相应的有机金属试剂(芳基锂/镁试剂),进一步的和葡萄糖酸内酯衍生物反应,构架药物分子骨架,随后通过脱保护基,甲基化,还原得到目标产物(us20060258749a1、wo2011039107a1);2.以碘代芳烃为原料,制备相应的有机金属试剂,进一步的和溴代葡萄糖衍生物反应,随后脱保护得到目标产物(cn105399735a,cn106905305a,石克金等."恩格列净合成工艺改进."中国医药工业杂志49.8(2018):4,d.yi,et al,org.lett.2018,20,7,1936
–
1940)。两种路线中,均涉及到将卤代芳烃制备为相应的有机金属试剂这一步骤,对反应温度,氧气,水分含量要求高。前者路线步骤繁琐,收率较低,整体效率较低,但是路线成熟,目前已被大规模应用于生产。后者为改进路线,具有步骤少的优势,但是原料仅限于碘代物,路线成本依旧很高,因此有待进一步改进。
4.光催化的还原偶联反应能直接实现两个亲电试剂之间的交叉偶联,从而构建碳碳键,反应条件温和,选择性好。但是目前的研究主要集中在理论研究部分,实际应用较少。2016年macmillan和雷爱文教授先后报道了光催化的还原偶联反应,实现了溴代芳烃和溴代烷烃之间的还原偶联反应,但是其底物范围存在一定的局限,官能团兼容性较差,对于氨基,羟基不能很好的兼容。故难以将其进一步的应用到药物合成当中(p.zhang,et al,j.am.chem.soc.2016,138,8084-8087;z.l.duan,et al,org.lett.2016,18,4012-4015)。基于此反应机理,本发明发展了恩/达格列净的光催化还原偶联合成法,步骤简单、成本低、原子经济性高、适宜于工业化生产。
技术实现要素:
5.针对现有方法的不足,本发明的目的在于提供了一种光催化的恩/达格列净前体及其合成方法,具有反应条件温和,操作简单,产率高,立体选择性好,副产物少,原料稳定易存储(没有使用活性较高的有机金属试剂),反应易于调控(无光照即反应停止),可大量制备等优点。
6.为了实现上述目的,本发明用以下技术措施:一种光催化的恩/达格列净前体的合成方法,包括如下步骤:
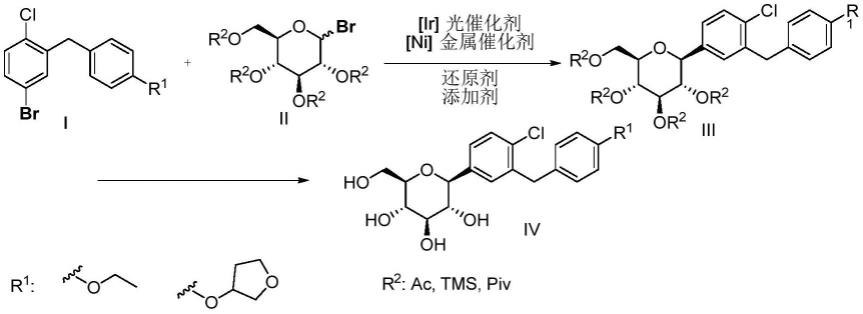
7.1)将溴代芳烃底物i、溴代葡萄糖底物ii、[ir]光催化剂、[ni]金属催化剂、还原剂、添加剂和极性溶剂混合后;在室温,光照,氮气氛围下反应,得到恩/达格列净的中间体iii;tlc监控反应进程,溴代芳烃底物i耗完毕即可停止反应,反应完成后,无需后处理,直接下一步反应;
[0008]
2)中间体iii在一定条件下脱去保护基,得到目标产物,随后柱层析分离纯化即可得到目标产物ⅳ。(r2为乙酰基或特戊酰基时,反应条件为:naoh,lioh,koh;r2为三甲基硅基时,反应条件为:柠檬酸,稀盐酸。r2优选为三甲基硅基保护基);
[0009]
合成路线如下:
[0010][0011]
其中,r1为乙氧基或者四氢呋喃-3-氧基;r2为乙酰基,三甲基硅基或特戊酰基。
[0012]
进一步地,[ir]光催化剂为ir(ppy)3,ir[df(cf3)ppy]2(dtbbpy)pf6或ir(ppy)2(dtbbpy)pf6,用量为0.1~2mol%(相对于底物i)。其结构如下图所示:
[0013][0014]
进一步地,[ni]金属催化剂为nicl2·
dtbbpy,nicl2,nibr2,ni(acac)2,ni(cod)2,用量为0.5~5mol%(相对于底物i)。
[0015]
进一步地,还原剂为三乙基硅烷或三乙胺,用量为1~2当量(相对于底物i)。
[0016]
进一步地,添加剂为氯化镁或氯化锌,用量为0~1当量(相对于底物i)。
[0017]
进一步地,溶剂为乙腈、甲苯、甲醇、dmf或dma;优选为dma。
[0018]
进一步地,溴代芳烃底物i和溴代葡萄糖底物ii的摩尔比为1:1~2。
[0019]
本发明提供了一种通过光催化溴代芳烃和溴代葡萄糖之间的还原交叉偶联制备恩/达格列净前体及其合成方法,具有反应条件温和,操作简单,产率高,立体选择性好,副产物少,原料稳定易存储(没有使用活性较高的有机金属试剂),反应易于调控(无光照即反应停止),可大量制备等优点。两步反应即可得到降血糖药物分子恩格列净(empagliflozin)和达格列净(dapagliflozin),两者结构如图所示。
[0020][0021]
与现有技术相比,本发明的技术方案具有如下有益效果:1.反应条件温和,操作简单,一锅两步法即可得到目标产物;2.产率高,立体选择性好,副产物少,可大量制备;3.同传统方法相比,原料稳定易存储,没有使用活性较高的有机金属试剂,降低了安全风险;4.反应安全可控,光照条件下才能反应,无光照即反应停止;5.反应机制新颖,当前类似的合成方法文献报道较少,还存在很大的优化空间。
附图说明
[0022]
图1为实施例1制备的达格列净氢谱图;
[0023]
图2为实施例1制备的达格列净碳谱图;
[0024]
图3为实施例3制备的恩格列净氢谱图;
[0025]
图4为实施例3制备的恩格列净碳谱图。
具体实施方式
[0026]
下面通过具体的实施案例进一步的描述本发明的内容,但并不限制本发明。
[0027]
实施例1
[0028]
以5-溴-2-氯-4
’‑
乙氧基二苯甲烷和2,3,4,6-四三甲基硅基-alpha-d-吡喃葡萄糖溴化物为原料合成达格列净中间体1a;随后在酸性条件下水解为达格列净。
[0029][0030]
在一个50ml schlenk管中,依次称取325.6mg(1mmol)5-溴-2-氯-4
’‑
乙氧基二苯甲烷,797.6mg(1.5mmol)2,3,4,6-四三甲基硅基-α-d-吡喃葡萄糖溴化物,9.8mg(0.01mmol)ir[df(cf3)ppy]2(dtbbpy)pf6,6.9mg(0.02mmol)nicl2·
dtbbpy,93.9mg(1mmol)mgcl2,随后加入磁子,盖上橡胶塞后,绑上氮气球。将管内置换为氮气后,加入303.3mg(3mmol)三乙胺和10ml dmf。将反应在420nm蓝光led灯照射下,室温搅拌反应6小时。随后向反应液中加入5ml 1n盐酸,搅拌反应两小时脱去tms保护基。反应结束后,调节ph至中性,柱层析分离纯化即可得到目标产物达格列净,产率60~73%(hplc纯度》99%)。图1为实施例1制备的达格列净氢谱图;图2为实施例1制备的达格列净碳谱图。
[0031]
实施例2
[0032]
以5-溴-2-氯-4
’‑
乙氧基二苯甲烷和2,3,4,6-四乙酰基-α-d-吡喃葡萄糖溴化物为原料合成达格列净中间体2a;随后在碱性条件下水解为达格列净。
[0033][0034]
在一个50ml schlenk管中,依次称取325.6mg(1mmol)5-溴-2-氯-4
’‑
乙氧基二苯甲烷,616.8mg(1.5mmol)2,3,4,6-四乙酰基-α-d-吡喃葡萄糖溴化物,9.8mg(0.01mmol)ir[df(cf3)ppy]2(dtbbpy)pf6,6.9mg(0.02mmol)nicl2·
dtbbpy,93.9mg(1mmol)mgcl2,随后加入磁子,盖上橡胶塞后,绑上氮气球。将管内置换为氮气后,加入303.3mg(3mmol)三乙胺和10ml dmf。将反应在420nm蓝光led灯照射下,室温搅拌反应6小时。随后向反应液中加入210.0mg(5mmol)lioh
·
h2o,搅拌反应两小时脱去tms保护基。反应结束后,调节ph至中性,柱层析分离纯化即可得到目标产物达格列净,产率55~63%(hplc纯度》99%)。
[0035]
实施例3
[0036]
以(3s)-3-[4-[(2-氯-5-溴苯基)甲基]苯氧基]四氢呋喃和2,3,4,6-四三甲基硅基-alpha-d-吡喃葡萄糖溴化物为原料合成恩格列净中间体3a;在酸性条件下水解为恩格列净。
[0037][0038]
在一个50ml schlenk管中,依次称取364.0mg(1mmol)
[0039]
(3s)-3-[4-[(2-氯-5-溴苯基)甲基]苯氧基]四氢呋喃,797.6mg(1.5mmol)2,3,4,6-四三甲基硅基-α-d-吡喃葡萄糖溴化物,9.8mg(0.01mmol)ir[df(cf3)ppy]2(dtbbpy)pf6,6.9mg(0.02mmol)nicl2·
dtbbpy,93.9mg(1mmol)mgcl2,随后加入磁子,盖上橡胶塞后,绑上氮气球。将管内置换为氮气后,加入303.3mg(3mmol)三乙胺和10ml dmf。将反应在420nm蓝光led灯照射下,室温搅拌反应6小时。随后向反应液中加入5ml 1n盐酸,搅拌反应两小时脱去tms保护基。反应结束后,调节ph至中性,柱层析分离纯化即可得到目标产物恩格列净,产率60~73%(hplc纯度》99%)。图3为实施例3制备的达格列净氢谱图;图4为实施例3制备的达格列净碳谱图。
[0040]
实施例4
[0041]
在一个250ml schlenk瓶中,依次称取3.3g(10mmol)5-溴-2-氯-4
’‑
乙氧基二苯甲烷,8.0g(15mmol)2,3,4,6-四三甲基硅基-α-d-吡喃葡萄糖溴化物,49.0mg(0.05mmol)ir[df(cf3)ppy]2(dtbbpy)pf6,69mg(0.2mmol)nicl2·
dtbbpy,0.94g(10mmol)mgcl2,随后加入磁子,盖上橡胶塞后,绑上氮气球。将反应瓶内置换为氮气后,加入3.0g(30mmol)三乙胺和100ml dmf。将反应在420nm蓝光led灯照射下,室温搅拌反应24小时。随后向反应液中加入50ml 1n盐酸,搅拌反应两小时脱去tms保护基。反应结束后,调节ph至中性,柱层析分离纯化即可得到目标产物达格列净,产率60~73%(hplc纯度》99%)。
[0042]
以上所述是本发明的优选实施方式而已,当然不能以此来限定本发明之权利范围,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和变动,这些改进和变动也视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1