一锅法制备莫匹拉韦中间体的方法与流程
1.本发明属于医药合成技术领域,具体涉及一种一锅法制备莫匹拉韦中间体 ((3ar,4r,6r,6ar)-6-(4-(羟氨)-2-恶嘧啶-1(2h)-甲基)-2,2-二甲基四氢呋喃[3,4-d][1,3] 二氧醇-4-基)异丁酸甲酯(化合物1)的方法。
背景技术:
[0002]
莫匹拉韦是一种核苷类药物,血浆酯酶代谢物通过干扰rna的合成而起作用,其代谢物造成病毒基因合成的高突变率,最终导致病毒死亡。由于rna聚合酶在rna病毒中结构相对保守,而该药物正是作用在rna聚合酶,因此可对多种变异株有效。经研究,抑制sars-cov-2 复制的活性比瑞德西韦高3-10倍,在多个临床前的sars-cov-2病毒感染预防、治疗和预防传播模型中均显示了活性。
[0003]
2021年8月9日,美国默沙东公司的口服抗新冠药物莫匹拉韦被澳大利亚药品管理局授予临时批准,也是全球首个获批的口服抗新冠药物。2021年11月,莫匹拉韦口服药在英国获批上市,用于治疗重症和住院风险较高的轻至中度covid-19成人患者。12月23日,美国食药监局(fda)批准了新冠口服药莫匹拉韦的紧急使用,用于18岁及以上治疗轻度到中度新冠肺炎。 2022年3月23日,韩国食品医药品安全处决定批准默沙东新冠口服药莫匹拉韦的紧急使用授权。
[0004]
((3ar,4r,6r,6ar)-6-(4-(羟氨)-2-恶嘧啶-1(2h)-甲基)-2,2-二甲基四氢呋喃 [3,4-d][1,3]二氧醇-4-基)异丁酸甲酯是抗病毒药物莫匹拉韦的重要中间体(化合物1), cas号:2346620-55-9,分子式:c
16h23
n3o7,结构式如下:
[0005][0006]
文献v.gopalsamuthiram等a concise route to mk-4482(eidd-2801)from cytidine: part 2[j],synlett 2021,32(03):326-328,报道了以保护的胞苷为原料,两个步骤合成((3ar,4r,6r,6ar)-6-(4-(羟氨)-2-恶嘧啶-1(2h)-甲基)-2,2-二甲基四氢呋喃 [3,4-d][1,3]二氧醇-4-基)异丁酸甲酯,以乙腈为溶剂,dbu为碱,反应液纯度35%;反应后处理用二氯甲烷进行柱层析,然后再用5%甲醇的氯仿溶液进行精制得到收率78%的中间体。下个反应步骤以70%异丙醇-水为溶剂反应,硫酸羟胺用量为3.2eq,72-73℃反应17小时,反应完毕浓缩除去溶剂,加入乙腈后过滤除去过量的硫酸羟胺,滤液浓缩后得到粗品;加入甲苯共沸除水析晶得到白色固体,纯度94%,收率96%。该方法存在几个问题:(1)第一个反应步骤中以昂贵的dbu为碱;(2)反应液纯度低只有35%,后处理还需要进行柱层析操作,然后再用混合溶剂进行精制提纯。(3)第二个步骤反应用到过量的硫酸羟胺,需要乙腈
溶解产品、再过滤除去不溶物盐;(4)用甲苯共沸除水,再处理得到的产品纯度和收率都不高。 (5)过量的硫酸羟胺受热不稳定,生产放大不安全;(6)反应溶剂不能实现回收和套用。
[0007]
专利wo2019113462(中国同族公开号cn111372592a,公开日:2020.07.03)公开了以保护的胞苷为原料,三个步骤合成((3ar,4r,6r,6ar)-6-(4-(羟氨)-2-恶嘧啶-1(2h)-甲基)-2,2-二甲基四氢呋喃[3,4-d][1,3]二氧醇-4-基)异丁酸甲酯,第一个步骤用异丁酸酐进行酰化反应,反应完毕浓缩、加入600ml乙酸乙酯溶解,用碳酸氢钠溶液、水和盐水分别洗涤两次,有机物用硫酸钠干燥,浓缩得到无色油。第二步骤以乙腈为反应溶剂,加入1,2, 4-三唑和n,n-二乙基乙胺搅拌溶解后,降温至0℃再加入三氯氧磷在氩气条件下反应,加水终止反应、浓缩、用二氯甲烷萃取洗涤、浓缩等,经过硅胶层析浓缩后得到固体;第三步骤以异丙醇溶解中间体,加入羟胺反应,反应完毕在环境温度下和45℃高真空除去一些溶剂,用乙酸乙酯溶解,溶液洗涤后干燥剂干燥、浓缩得到油进行结晶;过滤后的固体用乙醚洗涤,得到白色固体产物。该方法存在几个问题:(1)第一个步骤反应完毕浓缩、溶解、多个水溶液反复洗涤,操作繁琐;(2)第二个步骤需要用到三氯氧磷,产生的废水难以进行环保生化处理;(3)后处理用到硅胶层析操作进行提纯;(4)第三个步骤需要高真空浓缩;(5)溶剂萃取洗涤后,还需要用乙醚进行洗涤除杂,操作繁琐。(6)反应中用到乙酸乙酯、乙腈、二氯甲烷、异丙醇、乙醚等溶剂种类多,不利于回收和套用,导致原料成本高。
[0008]
总体而言,现有((3ar,4r,6r,6ar)-6-(4-(羟氨)-2-恶嘧啶-1(2h)-甲基)-2,2-二甲基四氢呋喃[3,4-d][1,3]二氧醇-4-基)异丁酸甲酯的制备方法,在一定程度上限制了本项目的推广应用。
技术实现要素:
[0009]
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。
[0010]
发明人在现有技术的基础上,针对上述的技术问题进行研究,提供了一种一锅法制备莫匹拉韦中间体的方法。该方法的合成路线操作简单、简化了反应后处理步骤、减少了溶剂种类的使用,去除了多步骤柱层析的精制,溶剂可以实现回收套用;原料成本和生产成本低、产生母液量少、生产更加绿色环保,所得莫匹拉韦中间体液相纯度和收率高,更适合于工业化大生产。
[0011]
本发明是通过下述的技术方案来实现的:
[0012]
一种一锅法制备莫匹拉韦中间体的方法,包括如下步骤:
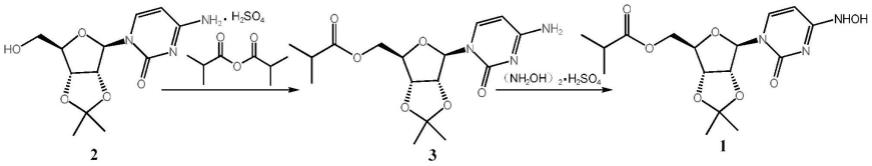
[0013][0014]
其中,结构式下面标注2的代表化合物2,结构式下面标注3的代表化合物3,结构式下面标注1的代表化合物1。
[0015]
上述的一锅法制备莫匹拉韦中间体的方法,包括如下步骤:
[0016]
s1:向反应釜中打入溶剂,搅拌下加入化合物2、dmap和有机碱,控温10-50℃加入异丁酸酐,反应完毕加入水,搅拌均匀得到化合物3的溶液;
[0017]
s2:向化合物3溶液中加入硫酸羟胺和无水醋酸钠,升温20-90℃反应至化合物3反应完毕,降温析晶,离心、湿品烘干得到中间体((3ar,4r,6r,6ar)-6-(4-(羟氨)-2-恶嘧啶
ꢀ‑
1(2h)-甲基)-2,2-二甲基四氢呋喃[3,4-d][1,3]二氧醇-4-基)异丁酸甲酯(化合物1)。
[0018]
上述的一锅法制备莫匹拉韦中间体的方法中,所述溶剂为二氯甲烷、1,2-二氯乙烷、正己烷、环己烷中的一种或者多种。
[0019]
优选的,所述溶剂为二氯甲烷,所述二氯甲烷和化合物2的质量比为2.0-8.0:1;优选为3.0-6.0:1。
[0020]
所述有机碱为二乙胺、三乙胺、二异丙基乙胺中的一种或者多种。
[0021]
优选的,所述有机碱为三乙胺。所述三乙胺和化合物2的质量比为0.40-1.33:1,优选为0.5-0.9:1。
[0022]
所述加入异丁酸酐的温度为20-40℃,所述升温反应至化合物3反应完毕,反应温度为 30-85℃。
[0023]
所述硫酸羟胺的加入量与化合物2的质量比为0.43-1.29:1,所述无水醋酸钠的加入量与化合物2的质量比为0.11-0.32:1。
[0024]
所述水和化合物2的质量比为3.0-8.0:1;优选为4.0-6.0:1。
[0025]
一种莫匹拉韦的制备方法,包括如下步骤:
[0026][0027][0028]
上述的莫匹拉韦的制备方法,包括如下步骤:
[0029]
(1)中间体((3ar,4r,6r,6ar)-6-(4-(羟氨)-2-恶嘧啶-1(2h)-甲基)-2,2-二甲基四氢呋喃[3,4-d][1,3]二氧醇-4-基)异丁酸甲酯(化合物1)的制备;
[0030]
s1:向反应釜中打入溶剂,搅拌下加入化合物2、dmap和有机碱,控温10-50℃加入异丁酸酐,反应完毕加入水,搅拌均匀得到化合物3的溶液;s2:向化合物3溶液中加入硫酸羟胺和无水醋酸钠,升温20-90℃反应至化合物3反应完毕,降温析晶,离心、湿品烘干得到中间体((3ar,4r,6r,6ar)-6-(4-(羟氨)-2-恶嘧啶-1(2h)-甲基)-2,2-二甲基四氢呋喃 [3,4-d][1,3]二氧醇-4-基)异丁酸甲酯(化合物1);
[0031]
(2)莫匹拉韦的制备
[0032]
向反应釜中打入二氯甲烷,加入化合物1,降温至-5-0℃,滴加浓盐酸,滴加完毕后保温反应3小时;化合物1反应完成后,控制温度0-5℃加入去离子水,然后缓慢滴加质量浓度26%氨水,调节溶液ph值至7.5左右,复测ph值不变后关停搅拌,静置半小时;分液,水相用乙酸乙酯萃取两次,合并有机相,用饱和食盐水洗涤,分液,浓缩有机相后加入去离子水,
加热溶解、活性炭脱色、过滤,然后缓慢降温至0-10℃析晶,离心,湿品烘干得到莫匹拉韦。
[0033]
上述的莫匹拉韦的制备方法中,所述二氯甲烷的用量与化合物1的质量比为3.0-8.0:1。
[0034]
所述水的用量与化合物1的质量比为2.0-5.0:1。
[0035]
所述乙酸乙酯萃取两次时,第一次萃取时乙酸乙酯的用量是化合物1质量的6倍,第二次萃取时乙酸乙酯的用量是化合物1质量的4倍。
[0036]
本发明的技术方案中,所述浓盐酸为质量百分比浓度为36%-38%的盐酸溶液。dmap为4
‑ꢀ
二甲氨基吡啶的简称。
[0037]
有益技术效果:
[0038]
(1)本发明的一锅法制备莫匹拉韦中间体的方法,合成路线操作简单、简化了反应后处理步骤、减少了溶剂种类的使用,原料成本和生产成本低、产生母液量少、生产更加绿色环保,所得莫匹拉韦中间体((3ar,4r,6r,6ar)-6-(4-(羟氨)-2-恶嘧啶-1(2h)-甲基)-2,2-二甲基四氢呋喃[3,4-d][1,3]二氧醇-4-基)异丁酸甲酯液相纯度和收率高,更适合于工业化大生产。产出的下步骤莫匹拉韦液相纯度达到99.9%以上,收率达到78.9%。
[0039]
(2)本发明的一锅法制备莫匹拉韦中间体的方法,在s1中,用和水不互溶的有机溶剂进行反应,解决了溶剂乙腈含水量高难以回收和套用的情况。本技术反应完毕加水直接进行下步反应,去除了有机相浓缩溶剂和洗涤去除有机盐的操作。
[0040]
(3)本发明的一锅法制备莫匹拉韦中间体的方法,化合物3无需单独浓缩分离,后续的升温反应中可以蒸馏几乎完全除去了有机溶剂;同时,蒸除有机溶剂后,加入的水再作为反应溶剂,直接去除了文献中溶剂异丙醇或者乙腈的使用。
[0041]
(4)本发明的一锅法制备莫匹拉韦中间体的方法,在由化合物3合成化合物1的步骤中,升温开始反应,同时随着反应溶液温度的升高,并开始蒸馏低沸点有机溶剂;缩短了反应时间,并缩短了本步骤的生产周期;减少了操作步骤,降低了生产成本。
[0042]
(5)本发明的一锅法制备莫匹拉韦中间体的方法,在由化合物3合成化合物1的步骤中,以合成化合物3步骤所用的有机溶剂和水为反应溶剂,反应中蒸馏出的有机溶剂,用去离子水洗涤后干燥除水,可以套用到下批次的步骤s1中;降低了产品原料成本,有利于工业化规模生产推广。
[0043]
(6)本发明的一锅法制备莫匹拉韦中间体的方法,在由化合物3合成化合物1中,因为硫酸羟胺的不稳定性,需要增加水量、降低溶液中硫酸羟胺的含量;本技术在化合物3步骤蒸除有机溶剂时,水量几乎不减少;因此,确保了反应溶液安全性。同时,相比文献选定的与水互溶的溶剂如乙腈或者异丙醇,提高了有机溶剂的回收率。
[0044]
(7)本发明的一锅法制备莫匹拉韦中间体的方法,用价廉的三乙胺为碱;同时去除了现有文献乙腈、乙酸乙酯、乙醚等溶剂的使用,去除了多步骤的各水溶液洗涤操作;也去除了柱层析操作,去除了文献中5%甲醇的氯仿溶液精制步骤。使产品原料成本大幅降低,并减少了废液的排放,绿色环保;也解决了操作繁琐的问题。
[0045]
(8)本发明的一锅法制备莫匹拉韦中间体的方法,液相纯度99.1%以上;而且为下步骤产出高质量的莫匹拉韦奠定了基础,实施例中产出的下步骤莫匹拉韦液相纯度达到99.9%以上,满足了单杂小于0.1%的质量要求。
具体实施方式
[0046]
下面结合具体实施方式来对本发明作更进一步的说明,以便本领域的技术人员更了解本发明,但并不以此限制本发明。
[0047]
实施例1:
[0048]
氮气保护下,向500l反应釜中打入105kg正己烷,加入35kg化合物2、0.2kgdmap、23.5kg 三乙胺,搅拌均匀。控温10-40℃加入22kg异丁酸酐,检测化合物2反应完毕;加入150kg 去离子水,搅拌下加入6.2kg无水醋酸钠和23.7kg硫酸羟胺。缓慢升温至正己烷蒸馏完全,再继续升温至70-80℃保温反应至化合物3反应完全。降温至20-30℃离心。湿品用30kg去离子水淋洗,再次离心的湿品烘干后得化合物1干品28.5kg,摩尔收率83.6%,液相纯度 99.14%。
[0049]
溶剂回收方案:
[0050]
s2中蒸馏回收的正己烷用水洗、分液,有机相干燥后,套用到下批次化合物2反应中。
[0051]
实施例2:
[0052]
氮气保护下,向5000l反应釜中打入1050kg二氯甲烷,加入350kg化合物2、2.2kgdmap、 235kg三乙胺,搅拌均匀。控温10-40℃加入220kg异丁酸酐,检测化合物2反应完毕;加入 1500kg去离子水,搅拌下加入62kg无水醋酸钠和237kg硫酸羟胺。缓慢升温至二氯甲烷蒸馏完全,再继续升温至70-80℃保温反应至化合物3反应完全。降温至20-30℃离心。湿品用 300kg去离子水淋洗,再次离心的湿品烘干后得化合物1干品291.7kg,摩尔收率85.6%,液相纯度99.35%。
[0053]
溶剂回收方案:
[0054]
s2中蒸馏回收的二氯甲烷用水洗、分液,有机相干燥后,套用到下批次化合物2反应中。
[0055]
实施例3:
[0056]
氮气保护下,向5000l反应釜中打入1,2-二氯乙烷1050kg,加入350kg化合物2、 2.2kgdmap、235kg三乙胺,搅拌均匀。控温10-40℃加入220kg异丁酸酐,检测化合物2反应完毕;加入1500kg去离子水,搅拌下加入62kg无水醋酸钠和237kg硫酸羟胺。缓慢升温至溶剂蒸馏完全,并且化合物3反应完全。降温至20-30℃离心。湿品用300kg去离子水淋洗,再次离心的湿品烘干后得化合物1干品277.7kg,摩尔收率81.5%,液相纯度99.52%。
[0057]
溶剂回收方案:
[0058]
s2中蒸馏回收的1,2-二氯乙烷用水洗、分液,有机相干燥后,套用到下批次化合物2 反应中。
[0059]
实施例4(回收溶剂的套用):
[0060]
氮气保护下,向5000l反应釜中打入实施例2回收的900kg二氯甲烷和150kg新的二氯甲烷,加入350kg化合物2、2.2kgdmap、235kg三乙胺,搅拌均匀。控温10-40℃加入220kg 异丁酸酐,检测化合物2反应完毕;加入1500kg去离子水,搅拌下加入62kg无水醋酸钠和 237kg硫酸羟胺。缓慢升温至二氯甲烷蒸馏完全,再继续升温至70-80℃保温反应至化合物3 反应完全。降温至20-30℃离心。湿品用300kg去离子水淋洗,再次离心的湿品烘干后得化合物1干品290.0kg,摩尔收率85.1%,液相纯度99.38%。
[0061]
溶剂回收方案:
[0062]
s2中蒸馏回收的二氯甲烷用水洗、分液,有机相干燥后,套用到下批次化合物2反应中。
[0063]
实施例5:莫匹拉韦的制备:
[0064]
1、反应式:
[0065][0066]
2、反应操作:
[0067]
向反应釜中打入二氯甲烷2000kg,加入400kg化合物1,搅拌降温至-5-0℃,滴加浓盐酸216kg,滴加完毕后保温反应3小时;化合物1反应完成后,控制温度0-5℃加入400kg去离子水,然后缓慢滴加质量浓度26%氨水170kg,调节溶液ph值至7.5左右,复测ph值不变后关停搅拌,静置半小时。分液,水相用乙酸乙酯2400kg和1600kg萃取两次,合并有机相,用饱和食盐水洗涤。分液,浓缩有机相后加入去离子水800kg,加热溶解、活性炭脱色、过滤,然后缓慢降温至0-10℃析晶2小时,离心,湿品烘干得到莫匹拉韦281.4kg,纯度99.92%,摩尔收率78.9%。
[0068]
以上实施例是为了说明本发明公开的实施方案,并不能理解为对本发明的限制。此外,本文所列出的各种修改以及发明中方法、组合物的变化,在不脱离本发明的范围和精神的前提下对本领域内的技术人员来说是显而易见的。虽然已结合本发明的多种具体优选实施例对本发明进行了具体的描述,但应当理解,本发明不应仅限于这些具体实施例。事实上,各种如上所述的对本领域内的技术人员来说显而易见的修改来获取发明都应包括在本发明的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1