一种合成吡咯烷衍生物的方法与流程
1.本发明涉及化学化工技术领域,特别是一种合成吡咯烷衍生物的方法。
背景技术:
2.1h-3-吡咯烷酮类化合物这种杂环片断是一种在天然产物、药物和生物活性组分中常见的结构,比如抗肿瘤药物喜树碱(camptothecin)的重要中间体,其衍生物广泛应用于精细化学品的合成,是合成杀虫剂、抗菌药等农药、医药的中间体。
3.现有技术中有数条合成路线可以用于1h-3-吡咯烷酮的合成,
4.如图2所示,路线1:报道了保护的3-羟基吡咯烷在氧化剂作用下,羟基氧化为羰基,然后脱保护得到1h-3-吡咯烷酮;工艺过程涉及到氧化剂,有放大生产的危险性或条件苛刻,同时3-羟基吡咯烷的成本也较高,难于工业化应用;
5.如图3所示,路线2:报道了苄胺和1-溴-2-酮-3-丁烯的合环来制备1h-3-吡咯烷酮,但收率极低,该路线较为简洁,但是1-溴-2-酮-3-丁烯的合成制备较为困难,同时合环反应的收率,低至20-30%,也不适合工业化生产。
6.如图4所示,路线3:报道了甘氨酸酯与丙烯酸酯经迈克尔加成,然后经保护伯胺,然后在碱的条件下合环,经加热脱羧,最后脱保护基得到1h-3-吡咯烷酮。该方法步骤长,总收率依然较低。该路线是现在较为成熟的制备路线,原料易得,总物料成本相对合适,但是制备路线达到5步,耗时较长,使得放大生产的总成本也是居高不下,同时,工艺中步骤长,涉及大量溶剂的使用和废水的排放,对环境保护不利。综上所述,现有的方法大多条件苛刻,或者反应过程由于放热等因素难以控制,导致工业化放大受阻。经过发明人长期研究,发明了一种合成吡咯烷衍生物的方法。
技术实现要素:
7.本发明的目的在于克服现有技术的缺点,提供一种合成吡咯烷衍生物的方法。
8.本发明的目的通过以下技术方案来实现:一种合成吡咯烷衍生物的方法,包括以下步骤:
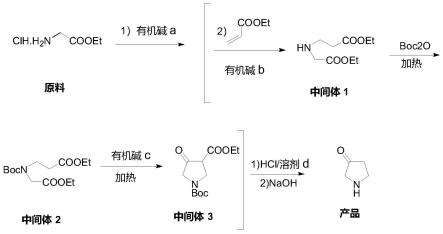
9.s1:将甘氨酸乙酯盐酸盐通有机碱a游离、过滤,浓缩得到液态的甘氨酸乙酯,加入丙烯酸酯,在混合物中,加入有机碱b,进行无溶剂的迈克尔加成,得到迈克尔加成产物,然后直接加入boc2o进行保护,(研磨)无溶剂加热,再加入有机碱c,继续(研磨)无溶剂并加热关环反应,得到中间体3;
10.s2:将中间体3与溶剂d混合加入酸然后加热,进行脱羧和脱保护反应,结束后,所得反应液浓缩,加入醇溶剂打浆,降温析出产品为吡咯烷酮的盐。
11.优选的,有机碱a为三乙胺、二异丙基乙基胺、dbu、乙醇钠和甲醇钠的一种或几种。
12.优选的,有机碱b为三乙胺、二异丙基乙基胺、dbu、乙醇钠、甲醇钠的一种或几种。
13.优选的,有机碱c为乙醇钠、甲醇钠、叔丁醇钠、叔丁醇钾的一种或几种。
14.优选的,无溶剂迈克尔反应的温度为25-85℃。
15.优选的,无溶剂boc2o保护仲胺的反应温度为30-80℃。
16.优选的,无溶剂加热关环反应的温度为45-90℃。
17.优选的,脱羧反应溶剂d为醇类溶剂与水的混合溶剂,醇类溶剂为:甲醇、乙醇、丙醇、异丙醇的一种或几种,且醇与水的比例为1:1至1:10。
18.优选的,步骤s2中加入的无机酸为盐酸、硫酸、硝酸中的一种或几种,加热脱羧温度为40-100℃。
19.本发明具有以下优点:本发明通过采用无溶剂条件,依次加入相应的物料,在不同的反应温度下反应,且不用分离,连续一锅进行,中间体3在酸性环境下,脱羧和脱boc同时进行,经浓缩后,加醇溶液打浆,所得有机物结晶析出,得到目标产物3-吡咯烷酮的盐,可以进一步纯化,该方法减少了90%的反应用有机溶剂,节省60%以上的工艺时间,总的生产成本降低50%以上,具有较明显的生产优势,并且对环境更友好,符合绿色化学理念。
附图说明
20.图1为合成吡咯烷衍生物方法的结构示意图;
21.图2为现有合成路线1的结构示意图;
22.图3为现有合成路线2的结构示意图;
23.图4为现有合成路线3的结构示意图;
24.图5为中间体3的核磁氢谱的示意图;
25.图6为最终产品吡咯烷酮的盐酸盐的核磁氢谱的示意图;
具体实施方式
26.为使本发明实施方式的目的、技术方案和优点更加清楚,下面将结合本发明实施方式中的附图,对本发明实施方式中的技术方案进行清楚、完整地描述,显然,所描述的实施方式是本发明一部分实施方式,而不是全部的实施方式。通常在此处附图中描述和示出的本发明实施方式的组件可以以各种不同的配置来布置和设计。
27.因此,以下对在附图中提供的本发明的实施方式的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施方式。基于本发明中的实施方式,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施方式,都属于本发明保护的范围。
28.需要说明的是,在不冲突的情况下,本发明中的实施方式及实施方式中的特征可以相互组合。
29.应注意到:相似的标号和字母在下面的附图中表示类似项,因此,一旦某一项在一个附图中被定义,则在随后的附图中不需要对其进行进一步定义和解释。
30.在本发明的描述中,还需要说明的是,除非另有明确的规定和限定,术语“设置”、“安装”、“相连”、“连接”应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或一体地连接;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通。对于本领域的普通技术人员而言,可以具体情况理解上述术语在本发明中的具体含义。
31.在本实施例中,如图1和图5所示,一种合成吡咯烷衍生物的方法,包括以下步骤:
32.s1:将甘氨酸乙酯盐酸盐通有机碱a游离、过滤,浓缩得到液态的甘氨酸乙酯,具体地说,在10l三口瓶内加入无水乙醇(6.50kg),室温下,将甘氨酸乙酯盐酸盐(2.7kg),加入至瓶内,控温20-30℃内,分批加入乙醇钠1.6kg,约20min加毕,搅拌3小时,过滤去除生成的无机物,40℃内,减压浓缩掉乙醇,得到油状物1.69kg,收率85%,即游离的甘氨酸乙酯;也可以采用在10l三口瓶内,加入四氢呋喃(6.0kg),室温下,将甘氨酸乙酯盐酸盐(2.0kg),加入至瓶内,控温20-30℃内,滴加三乙胺1.6kg,约20min加毕,搅拌3小时,过滤去除生成的三乙胺盐酸盐,40℃内,减压浓缩掉四氢呋喃,得到油状物1.3kg,收率86.3%,即游离的甘氨酸乙酯。加入丙烯酸酯,在混合物中,加入有机碱b,进行无溶剂的迈克尔加成,得到迈克尔加成产物,然后直接加入boc2o进行保护,(研磨)无溶剂加热,再加入有机碱c,继续(研磨)无溶剂并加热关环反应,得到中间体3;优选的,加入boc2o进行保护,(研磨)无溶剂加热,2小时后,再加入有机碱c,继续(研磨)无溶剂并加热关环反应2小时,得到中间体3。具体地说,于5l三口瓶内,加入游离的甘氨酸乙酯1.5kg及丙烯酸乙酯1.45kg,室温下,加入二异丙基乙基胺0.7kg、粉状4a分子筛500g,加热至65-70℃,搅拌反应6小时,反应结束后,静止降温至0℃保持1小时,倾去上层清液,也就是二异丙基乙基胺,所得浅褐色油状液体,为中间体1,不用纯化直接加入boc2o2.3kg,搅拌加热至55-65℃,反应3小时,反应结束后,所得浅褐色油状液体,为中间体2,不用纯化直接加入乙醇钠固体0.7kg,搅拌加热至45-55℃时,放热明显,控温不超过90℃,1小时后,需加热至70℃继续反应3小时,反应结束后,所得深褐色油状液体,为中间体3。或者采用在5l三口瓶内,加入游离的甘氨酸乙酯1.2kg及丙烯酸乙酯1.2kg,室温下,加入dbu0.88kg、粉状4a分子筛300g,加热至45-50℃,搅拌反应10小时,反应结束后,静止降温至室温,所得浅褐色油状液体,为中间体1,不用纯化直接加入boc2o1.8kg,搅拌加热至45-55℃,反应5小时,反应结束后,所得浅褐色油状液体,为中间体2,不用纯化加入叔丁醇钾固体0.98kg,搅拌加热至45-55℃时,放热明显,控温不超过90℃,1小时后,需加热至60-80℃继续反应3小时,反应结束后所得深褐色油状液体,为中间体3。或者采用在3l三口瓶内,加入游离的甘氨酸乙酯1.0kg及丙烯酸乙酯0.97kg,室温下,加入甲醇钠0.52kg、粉状4a分子筛200g,加热至50-60℃,搅拌反应5小时,反应结束后,静止降温保持1小时,简单滤除下层固体物质,也就是分子筛和醇钠,所得浅褐色油状液体,为中间体1,不用纯化直接加入boc2o2.3kg,搅拌加热至40-50℃,反应7小时,反应结束后,所得浅褐色油状液体,为中间体2,不用纯化直接加入叔丁醇钠固体0.6kg,搅拌加热至60-70℃时,控温不超过90℃,1小时后,保温70-80℃继续反应3小时,反应结束后,所得深褐色油状液体,为中间体3。
33.s2:将中间体3与溶剂d混合加入酸然后加热,进行脱羧和脱保护反应,结束后,所得反应液浓缩,加入醇溶剂打浆,降温析出,过滤所得产品为吡咯烷酮的盐。该方法减少了90%的反应用有机溶剂,节省60%以上的工艺时间,总的生产成本降低50%以上,具有较明显的生产优势。具体地说,在10l三口瓶内预先加入浓盐酸(2.3kg),室温下,将油状物中间体3(2.0kg)于95%乙醇(2.0kg)溶液,加入三口瓶。缓慢升温35-45℃反应5小时,再次升温回流反应10小时,反应结束后,将反应液降温,控温45-55℃,减压浓缩去除大部分乙醇和水,降温至室温,加入异丙醇2kg,再次降温0-10℃,搅拌析晶4小时。过滤,滤液做废液处理,滤饼以乙酸乙酯1.0kg淋洗,再抽干,滤饼转至烘箱,在40-50℃减压烘干,得类白色固体产品0.35kg。或者在10l三口瓶内预先加入浓盐酸(2.3kg),室温下,将油状物中间体3(2.0kg)
于乙醇(2.0kg)溶液,加入三口瓶。缓慢升温45-55℃反应5小时,再次升温回流反应10小时,检测反应结束。将反应液降温,控温45-55℃,减压浓缩去除大部分乙醇和水,降温至室温,加入无水乙醇2kg,冰浴降温,控温15℃内,析出类白色固体过滤,并将产品置烘箱,减压烘干,可以得到类白色固体产品1h-3-吡咯烷酮盐酸盐0.30kg。
34.进一步的,有机碱a为三乙胺、二异丙基乙基胺、dbu、乙醇钠和甲醇钠的一种或几种。优选的,有机碱a为三乙胺。
35.再进一步的,有机碱b为三乙胺、二异丙基乙基胺、dbu、乙醇钠、甲醇钠的一种或几种。优选的,有机碱b为dbu。
36.在本实施例中,有机碱c为乙醇钠、甲醇钠、叔丁醇钠、叔丁醇钾的一种或几种。具体地说,有机碱c为叔丁醇钾。
37.进一步的,无溶剂迈克尔反应的温度为25-85℃。优选的,无溶剂迈克尔反应的温度为45-55℃。
38.再进一步的,无溶剂boc2o保护仲胺的反应温度为30-80℃。优选的,无溶剂boc2o保护仲胺的反应温度为45-55℃。
39.在本实施例中,无溶剂加热关环反应的温度为45-90℃。优选的,无溶剂加热关环反应的温度为60-70℃。
40.进一步的,溶剂d为醇类溶剂与水的混合溶剂,醇类溶剂为:甲醇、乙醇、丙醇、异丙醇的一种或几种,且醇与水的比例为1:1至1:10。具体地说,醇类溶剂为乙醇,其中醇与水的比例为1:4。
41.再进一步的,步骤s2中加入的无机酸为盐酸、硫酸、硝酸中的一种或几种,加热脱羧温度为40-100℃。具体地说,加入的酸为盐酸,加热脱羧温度为80-90℃。
42.尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1