一种2,3-辛二酮的制备方法与流程
1.本发明涉及一种2,3-辛二酮的制备方法,属于有机合成技术领域。
背景技术:
2.2,3-辛二酮(2,3-octanedione),cas:585-25-1,2,3-辛二酮是一种黄色油状并易挥发的液体,带有奶油香和油脂香,主要用于咖啡、烟草、焙烤食品、奶制品香精中,天然存在于轮叶冬青花精油、无花果挥发油等。目前2,3-辛二酮为我国gb2760-2011规定为可食用香料,编码s0292。2,3-辛二酮作为一种替代物,源于其性质稳定,有相似的香气,进而丰富该类食品香料。
3.文献[journal ofmolecular catalysis b:enzymatic,2013,vol.85-86,p.93-98]报道采用2,3-辛二酮合成手性3-羟基-3-甲基壬烷-2,4-二酮。(ss,rr)-构型已被公认为绿茶冲泡时挥发的主要风味成分之一。其化学式如下:
[0004][0005]
2,3-辛二酮合成的方法较多,其中[journal oforganic chemistry,1987,52,4851-4855]报道2,3-辛二醇与三氟乙酸酐“活化”二甲基亚砜形成氧化剂进行氧化合成。但2,3-辛二醇成本明显更高,不利于工业化合成。其反应方程式如下:
[0006][0007]
文献[journal of the chemical society.chemical communications,1987,692-693]报道1-已醛与醋酸酐在氯化钴催化下合成2,3-辛二酮,转化率高达98%,但原料价格较贵,分离较难(少量原料、2,3-丁二酮和醋酸与2,3-辛二酮不易分离),并且用到易制毒管控化合物醋酸酐,不利于工业化生产。其反应方程式如下:
[0008][0009]
cn103113198b发明专利公开了一种2,3-辛二酮的清洁生产方法,采用乙醛、正己醛和复合催化剂高压偶联,随后氧化得到2,3-辛二酮,总收率48%。
[0010]
传统工艺采用亚硝酸异戊酯和2-辛酮反应生成3-肟-2-辛酮,再水解成2,3-辛二酮,收率47%。本发明对传统工艺进行改进,第一步亚硝酸异戊酯改成亚硝酸叔丁酯(如:亚硝酸正丙酯、亚硝酸正丁酯和亚硝酸正戊酯为限制类或控制类化工用品,它们可以制成一种致幻rush poppers,其具有催情气体,其副作用极大,可能导致窒息,心律失常,心血管抑制,肝肾毒性,高铁血红蛋白血症,神经功能失调)、从无溶剂非均相反应改为有溶剂均相反
应、从减压蒸馏到石油醚析晶。第二步从亚硝酸钠与硫酸水解改为采用50%乙醛酸与硫酸或甲磺酸组合水解。总收率从47%提高到67-80%。
[0011]
本发明采用流程更简便,避免了精馏过程,对设备要求低,反应条件温和,总收率高的制备方法,适应其工厂生产,以满足日益增长的市场需求。
技术实现要素:
[0012]
为了克服上述技术缺陷,本发明对传统工艺进行改进,以2-辛酮为原料与亚硝酸叔丁酯进行亲核取代,随后在50%乙醛酸及催化量路易斯酸下水解得到2,3-辛二酮,本发明对传统工艺进行改进,使其流程简便,反应条件温和,无需精馏等特殊设备,其中母液及废酸可重复利用2-3次,总收率高达67-80%。
[0013]
本发明所述一种2,3-辛二酮的制备方法,反应方程式表示如下:
[0014][0015]
包括如下步骤:
[0016]
a):将2-辛酮为原料与催化量浓盐酸在有机溶剂中混合,控制温度35-40℃滴加亚硝酸叔丁酸,甲醇淬灭,通过后处理得到3-肟-2-辛酮;
[0017]
b):将50%乙醛酸和路易斯酸催化剂混合,加热下分批加入3-肟-2-辛酮,分层,减压蒸馏得到2,3-辛二酮。
[0018]
进一步地,在上述技术方案步骤a)中,所述2-辛二酮、浓盐酸与亚硝酸叔丁酯摩尔比为1:0.01-0.1:1.20-1.25。
[0019]
进一步地,在上述技术方案步骤a)中,所述有机溶剂选自四氢呋喃、异丙醚和甲基叔丁基醚。
[0020]
进一步地,在上述技术方案步骤a)中,所述后处理为加入石油醚,碳酸钠水溶液水洗ph=9-10,有机相10-25℃减压浓缩蒸除极性溶剂,加入石油醚降温至-10℃,大量固体析出,过滤得到3-肟-2-辛酮,母液可套用。
[0021]
进一步地,在上述技术方案步骤b)中,所述路易斯酸催化剂选自浓硫酸或甲磺酸。
[0022]
进一步地,在上述技术方案步骤b)中,所述3-肟-2-辛酮、50%乙醛酸与路易斯酸摩尔比为1:10-15:0.01-0.1。
[0023]
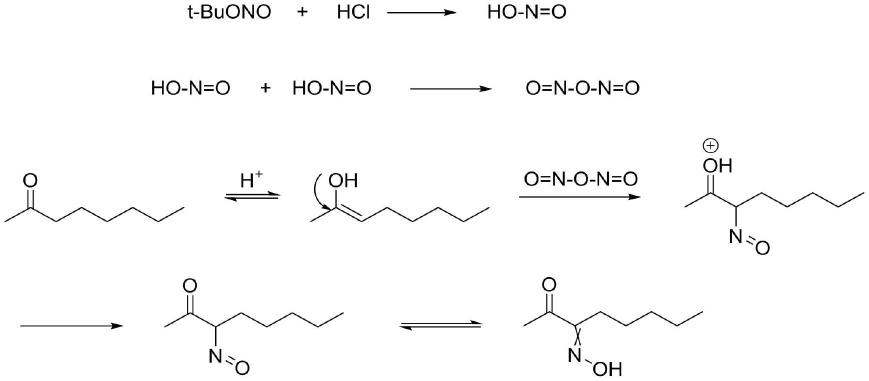
本发明第一步中,反应机理推测过程如下:
[0024][0025]
2-辛酮在酸性条件下生成烯醇式,同时亚硝酸叔丁酯在酸性条件下生成亚硝酸,进而生成n2o3,随后亲核取代α位,最后异构化得到3-肟-2-辛酮。
[0026]
发明有益效果
[0027]
1、第一步中选用硝化试剂亚硝酸叔丁酯,其活性高/价格低廉,官能团兼容性好,沸点相对较低,为非限制类或管控化工品,添加极性溶剂可增加其反应效果,提高转化率;并用石油醚固化,得到中间体质量高,便于最终产品纯化(无需精馏)。第二步选用50%乙醛酸水解,效果更好,更稳定,转化率大于99.5%,收率高达94%。
[0028]
2、产品2,3-辛二酮与原料2-辛酮沸点相当,分别是173.5℃和172.4℃,并且2,3-辛二酮更是易挥发,如用2-辛酮为原料合成,必然中间体要进行纯化,并且中间体3-肟-2-辛酮受热情况下会缓慢生成2,3-辛二酮,因此在合成中间体是选择溶剂沸点相对低,需要高真空减压浓缩蒸除溶剂。
[0029]
3、主要三废第一步母液和第二步酸水可套用2-3次,整体减少三废处理。
附图说明
[0030]
图1为实施例1中得到产品的核磁共振hnmr谱图;
[0031]
图2为实施例3中得到产品的核磁共振hnmr谱图。
具体实施例
[0032]
下面通过具体实例对本发明进行进一步说明。这些实施例应理解为仅用于说明本发明而不用于限制本发明的保护范围。在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等效变化和修改同样落入本发明权利要求所限定的范围。
[0033]
3-肟-2-辛酮的合成
[0034]
实施例1
[0035][0036]
氮气保护下,将2-辛酮128.2g(1eq,1.0mol)、150ml四氢呋喃和5ml浓盐酸加入反
应瓶中,室温下不间断滴加亚硝酸叔丁酯123.7g(1.2eq,1.2mol),滴加过程中放热,升温并保持温度在40
±
3℃。滴加结束反应1小时,gc检测原料剩余4-7%,加入少量石油醚约150ml,加入碳酸钠水洗两次,氯化钠饱和溶液洗,滤液15-25℃浓缩至几乎不流液,加入石油醚200ml降温至-10℃,大量固体析出,过滤,滤饼用冷石油醚漂洗,干燥得到3-肟-2-辛酮130.5g。gc 99.6%,收率83%。1hnmr(400mhz,cdcl3):8.48(s,1h),2.58-2.49(m,2h),2.32(s,3h),1.50-1.24(m,6h),0.90-0.81(m,3h).
[0037]
实施例2
[0038][0039]
氮气保护下,将2-辛酮128.2g(1eq,1.0mol)、150mlmtbe和5ml浓盐酸加入反应瓶中,室温下不间断滴加亚硝酸叔丁酯123.7g(1.2eq,1.2mol),滴加过程中放热,升温并保持温度在40
±
3℃。滴加结束,反应1小时,gc检测原料剩余7%,加入mtbe约200ml,加入碳酸钠水洗两次,氯化钠饱和溶液洗涤一次,滤液15-25℃浓缩至几乎不流液,石油醚200ml,降温至-10℃,大量固体析出,过滤,滤饼用冷的石油醚漂洗,产品干燥。得到3-肟-2-辛酮125.5g。gc 99.5%,收率79.8%。
[0040]
2,3-辛二酮的合成
[0041]
实施例3
[0042][0043]
在反应瓶内,将740.4g 50%乙醛酸(10eq,5mol)和4.9g浓硫酸(0.1eq,0.05mol)混合,升温至60-65℃,分批加入3-肟-2-辛酮78.6g(1eq,0.5mol),加毕保温反应8小时,中间体剩余约0.25%。分层,有机相加入碳酸钠水溶液洗涤,水洗,无水硫酸镁干燥,25-35℃高真空减压蒸馏得到2,3-辛二酮57.7g,gc 99.4%,收率81.2%。1hnmr(400mhz,cdcl3):2.75-2.72(m,2h),2.33(s,3h),1.62-1.55(m,2h),1.33-1.29(m,4h),0.91-0.89(m,3h).
[0044]
实施例4
[0045][0046]
在反应瓶内,将740.4g 50%乙醛酸(10eq,5mol)和4.8g甲磺酸(0.1eq,0.05mol)混合,升温至60-65℃,分批加入3-肟-2-辛酮78.6g(1eq,0.5mol),加毕保温反应10小时,中间体剩余约0.5%。分层,有机相加入碳酸钠水溶液洗涤,水洗,无水硫酸镁干燥,25-35℃高真空减压蒸馏得到2,3-辛二酮57.0g,gc 99.0%,收率80.1%。
[0047]
实施例5
[0048]
公斤级实验
[0049]
氮气保护下,30l玻璃夹套反应釜内加入2-辛酮6.411kg(50mol)加入6kg四氢呋喃和230ml浓盐酸,室温下不间断的滴加亚硝酸叔丁酯6.18kg(60mol),滴加过程中放热,升温
并保持温度在40
±
3℃,滴加结束后,反应1小时,gc检测原料剩余4%。加入50g甲醇淬灭,加入石油醚约6l,加入饱和碳酸钠水溶液1.5kg洗涤两次,氯化钠溶液洗,滤液15-25℃浓缩至几乎不流液,加入石油醚替换,加入石油醚12l,降温至-10℃(有大量固体析出),过滤,滤饼用冷石油醚漂洗(滤液用于下实施例6),产品干燥得到3-肟-2-辛酮6.509kg。gc 99.6%,收率82.8%。
[0050]
在100l搪瓷反应釜内,将61.3kg 50%乙醛酸(10eq,414mol)和40.6g浓硫酸(0.01eq,0.414mol)混合,升温至60-65℃,分批加入3-肟-2-辛酮6.509kg(1eq,414mol),控温65-70℃反应12小时,中间体剩余约0.21%。分层,水相用于实施例6,有机相加入碳酸钠水溶液洗涤,水洗,无水硫酸镁干燥,少量石油醚淋洗(用于实施例6),25-35℃高真空减压蒸馏得到2,3-辛二酮5.086kg,gc 99.6%,收率86.4%。
[0051]
实施例6
[0052]
公斤级母液和酸水套用实验
[0053]
氮气保护下,30l玻璃夹套反应釜内加入2-辛酮6.411kg(50mol)加入6kg四氢呋喃和230ml浓盐酸,室温下不间断的滴加亚硝酸叔丁酯6.18kg(60mol),滴加过程中放热,升温并保持温度在40℃
±
3,滴加结束后,反应1小时,gc检测原料剩余4.5%。加入50g甲醇淬灭,加入石油醚约6l,加入饱和碳酸钠水溶液1.5kg洗涤两次,氯化钠溶液洗,滤液15-25℃浓缩至几乎不流液,加入石油醚替换,加入实施例5中母液,降温至-10℃(有大量固体析出),过滤,滤饼用冷的石油醚漂洗,产品干燥得到3-肟-2-辛酮6.736kg。gc99.6%,收率85.7%。
[0054]
在100l搪瓷反应釜内,将上实施例5中水相、实施例5中石油醚淋洗液和40.6g浓硫酸(0.01eq,0.414mol)混合,升温至60-65℃,分批加入3-肟-2-辛酮6.736kg(1eq,428.5mol),控温65-70℃反应12小时,中间体剩余约0.2%。分层,有机相加入碳酸钠水溶液洗涤,水洗,无水硫酸镁干燥,少量石油醚淋洗,0℃减压蒸出石油醚,随后25-35℃高真空减压蒸馏得到2,3-辛二酮5.734kg,gc 99.1%,收率94.1%。
[0055]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1