一种艾沙康唑中间体III的制备方法与流程
一种艾沙康唑中间体iii的制备方法
技术领域
1.本发明涉及化学合成技术领域,尤其是一种艾沙康唑中间体iii的制备方法,具体为(z)-3-(2,5-二氟苯基)-4-(1h-1,2,4-三氮唑-1-基)丁-2-烯腈的制备方法。
背景技术:
2.近20年来,由于造血干细胞移植、实体器官移植、肿瘤化疗的广泛应用,侵袭性真菌感染的患病率呈显著上升趋势。侵袭性真菌感染主要由念珠菌属和曲霉菌属所致,其易感人群为免疫力低下患者,多发生在血液、icu、移植以及呼吸和感染领域,其中血液科病人,如骨髓移植、白血病和淋巴瘤患者的比重约占61%,感染和呼吸科患者约占17%,主要集中为艾滋病、呼吸衰竭和免疫功能低下的患者。
3.cn200680044974,公开了艾沙康唑中间体及工艺,经环氧化后再引入腈基。
4.现有艾沙康唑类药物合成方法中,基本都是先环氧化得到环氧乙烷,再设法引入腈基,且引入腈基的过程都使用了剧毒物料三甲基腈硅烷或丙酮氰醇。本发明提供了一种利用wittig反应引入氰基的方法,为了达到此目的特提供了含氰基化合物(式iii)的制备方法。
技术实现要素:
5.本发明的目的是:克服现有技术中的不足,提供一种艾沙康唑中间体iii的制备方法。
6.为实现上述目的,本发明中采用的技术方案如下:一种艾沙康唑中间体iii的制备方法,所述式iii的化学式如下:。
7.进一步的,所述制备方法包括以下步骤:以1-(2,5-二氟苯基)-2-(1h-1,2,4-三氮唑-1-基)乙酮(式ii)和2-卤代乙腈为原料,在适宜溶剂、温度条件下进行wittig反应,制得艾沙康唑中间体式iii所示化合物(z)-3-(2,5-二氟苯基)-4-(1h-1,2,4-三氮唑-1-基)丁-2-烯腈。
8.。
9.进一步的,所述wittig反应温度为10℃至50℃。
10.进一步的,所述wittig反应温度为20℃至30℃。
11.进一步的,所述wittig反应时间为5-12小时,
进一步的,所述wittig反应时间为6-8小时。
12.进一步的,所述制备中间体iii所用反应溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、四氢呋喃,优选为n,n-二甲基甲酰胺。
13.进一步的,所用物料卤代乙腈为氯代乙腈、溴代乙腈,优选为氯代乙腈。
14.进一步的,所述制备中间体iii的过程中,化合物1-(2,5-二氟苯基)-2-(1h-1,2,4-三氮唑-1-基)乙酮(式ii)与三苯基膦与卤代乙腈的摩尔比为1.0:(1.2-1.8):(1.2-1.8)。
15.进一步的,所述制备中间体iii的过程中,1-(2,5-二氟苯基)-2-(1h-1,2,4-三氮唑-1-基)乙酮(式ii)与三苯基膦与卤代乙腈的摩尔比为1.0:(1.5-1.6):(1.5-1.6)。
16.采用本发明中技术方案具有以下有益效果:用wittig反应引入了腈基制得的(z)-3-(2,5-二氟苯基)-4-(1h-1,2,4-三氮唑-1-基)丁-2-烯腈(式iii)可用于进一步制备艾沙康唑及艾沙康唑类关键中间体。
17.wittig反应引入了腈基的方法避免了艾沙康唑现有技术中使用剧毒物料三甲基腈硅烷或丙酮氰醇的步骤,对环境污染小。
18.本方法制备(z)-3-(2,5-二氟苯基)-4-(1h-1,2,4-三氮唑-1-基)丁-2-烯腈工艺简便,所得化合物iii纯度高,收率理想能实现工业化生产。
附图说明
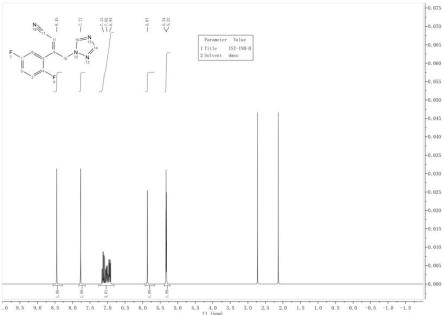
19.图1为艾沙康唑中间体iii的氢谱图。
实施方式
20.下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
21.实施例1:(z)-3-(2,5-二氟苯基)-4-(1h-1,2,4-三氮唑-1-基)丁-2-烯腈(式iii)的制备:反应瓶中加入196.7g(0.75mol)三苯基膦、800ml甲苯开启搅拌,再加入56.6g(0.75mol)2-氯乙腈,室温下搅拌15分钟至体系均匀,升温至110度回流5-6小时后自然降温至25度左右,析出大量白色固体,抽滤,滤饼用甲基叔丁基醚淋洗(100ml*3次),得类白色固体,加入800mldmf溶解后,氮气保护,待用。
22.另取干净反应瓶,氮气保护,依次加入1000ml的dmf、氢化钠18.0g(0.75mol),室温下搅拌均匀,继续在室温下滴加上述待用的三苯基膦dmf溶液,约2-3小时滴加完毕后再搅拌1小时,保持20度温度范围内滴加1-(2,5-二氟苯基)-2-(1h-1,2,4-三氮唑-1-基)乙酮(式ii)的dmf溶液[111.6g(0.5mol)式ii和500ml的dmf],滴加完毕后,继续保持20度进行wittig反应6小时,tlc中控,反应完全,加入500ml自来水搅拌萃灭30分钟,抽滤除去大部分三苯基氧磷,所得反应液用乙酸乙酯进行提取(600ml*5次),合并有机相,水洗(100ml*2次),饱和食盐水洗(100m*1次),脱溶后用300ml乙腈进行溶解后,再加入8.5g(0.023mol)(-)riboflavin,搅拌均匀,室温下在402nm的紫外灯环境下照射24小时,脱溶至干,200ml甲基叔丁基醚室温下打浆1小时,抽滤,烘干后得111.27g类白色固体,hplc纯度98.33%,摩尔
收率90.38%。取少量用甲基叔丁基醚做热打浆后,抽滤烘干,送检核磁氢谱检测,结果见图1。实施例2:(z)-3-(2,5-二氟苯基)-4-(1h-1,2,4-三氮唑-1-基)丁-2-烯腈(式iii)的制备:反应瓶中加入196.7g(0.75mol)三苯基膦、800ml甲苯开启搅拌,再加入56.6g(0.75mol)2-氯乙腈,室温下搅拌15分钟至体系均匀,升温至110度回流5-6小时后自然降温至25度左右,析出大量白色固体,抽滤,滤饼用甲基叔丁基醚淋洗(100ml*3次),得类白色固体,加入800mldmf溶解后,氮气保护,待用。
[0023]
另取干净反应瓶,氮气保护,依次加入1000ml的dmf、氢化钠18.0g(0.75mol),室温下搅拌均匀,继续在室温下滴加上述待用的三苯基膦dmf溶液,约2-3小时滴加完毕后再搅拌1小时,保持30度温度范围内滴加1-(2,5-二氟苯基)-2-(1h-1,2,4-三氮唑-1-基)乙酮(式ii)的dmf溶液[111.6g(0.5mol)式ii和500ml的dmf],滴加完毕后,继续保持30度进行wittig反应8小时,tlc中控,反应完全,加入500ml自来水搅拌萃灭30分钟,抽滤除去大部分三苯基氧磷,所得反应液用乙酸乙酯进行提取(600ml*5次),合并有机相,水洗(100ml*2次),饱和食盐水洗(100ml*1次),脱溶后用300ml乙腈进行溶解后,再加入8.5g(0.023mol)(-)riboflavin,搅拌均匀,室温下在402nm的紫外灯环境下照射24小时,脱溶至干,200ml甲基叔丁基醚室温下打浆1小时,抽滤,烘干后得112.34g类白色固体,hplc纯度97.56%,摩尔收率91.25%。
[0024]
实施例3:(z)-3-(2,5-二氟苯基)-4-(1h-1,2,4-三氮唑-1-基)丁-2-烯腈(式iii)的制备:反应瓶中加入209.8g(0.80mol)三苯基膦、800ml甲苯开启搅拌,再加入60.4g(0.80mol)2-氯乙腈,室温下搅拌15分钟至体系均匀,升温至110度回流5-6小时后自然降温至25度左右,析出大量白色固体,抽滤,滤饼用甲基叔丁基醚淋洗(100ml*3次),得类白色固体,加入800mldmf溶解后,氮气保护,待用。
[0025]
另取干净反应瓶,氮气保护,依次加入1000ml的dmf、氢化钠18.0g(0.75mol),室温下搅拌均匀,继续在室温下滴加上述待用的三苯基膦dmf溶液,约2-3小时滴加完毕后再搅拌1小时,保持30度温度范围内滴加1-(2,5-二氟苯基)-2-(1h-1,2,4-三氮唑-1-基)乙酮(式ii)的dmf溶液[111.6g(0.5mol)式ii和500ml的dmf],滴加完毕后,继续保持30度进行wittig反应8小时,tlc中控,反应完全,加入500ml自来水搅拌萃灭30分钟,抽滤除去大部分三苯基氧磷,所得反应液用乙酸乙酯进行提取(600ml*5次),合并有机相,水洗(100ml*2次),饱和食盐水洗(100ml*1次),脱溶后用300ml乙腈进行溶解后,再加入8.5g(0.023mol)(-)riboflavin,搅拌均匀,室温下在402nm的紫外灯环境下照射24小时,脱溶至干,200ml甲基叔丁基醚室温下打浆1小时,抽滤,烘干后得112.87g类白色固体,hplc纯度97.31%,摩尔收率91.68%。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1