结合PSMA的双模式放射性示踪剂及治疗剂
结合psma的双模式放射性示踪剂及治疗剂
1.本技术是申请号为202080024836.9、申请日为2020年1月30日、发明名称为“结合psma的双模式放射性示踪剂及治疗剂”的中国发明专利申请的分案申请。
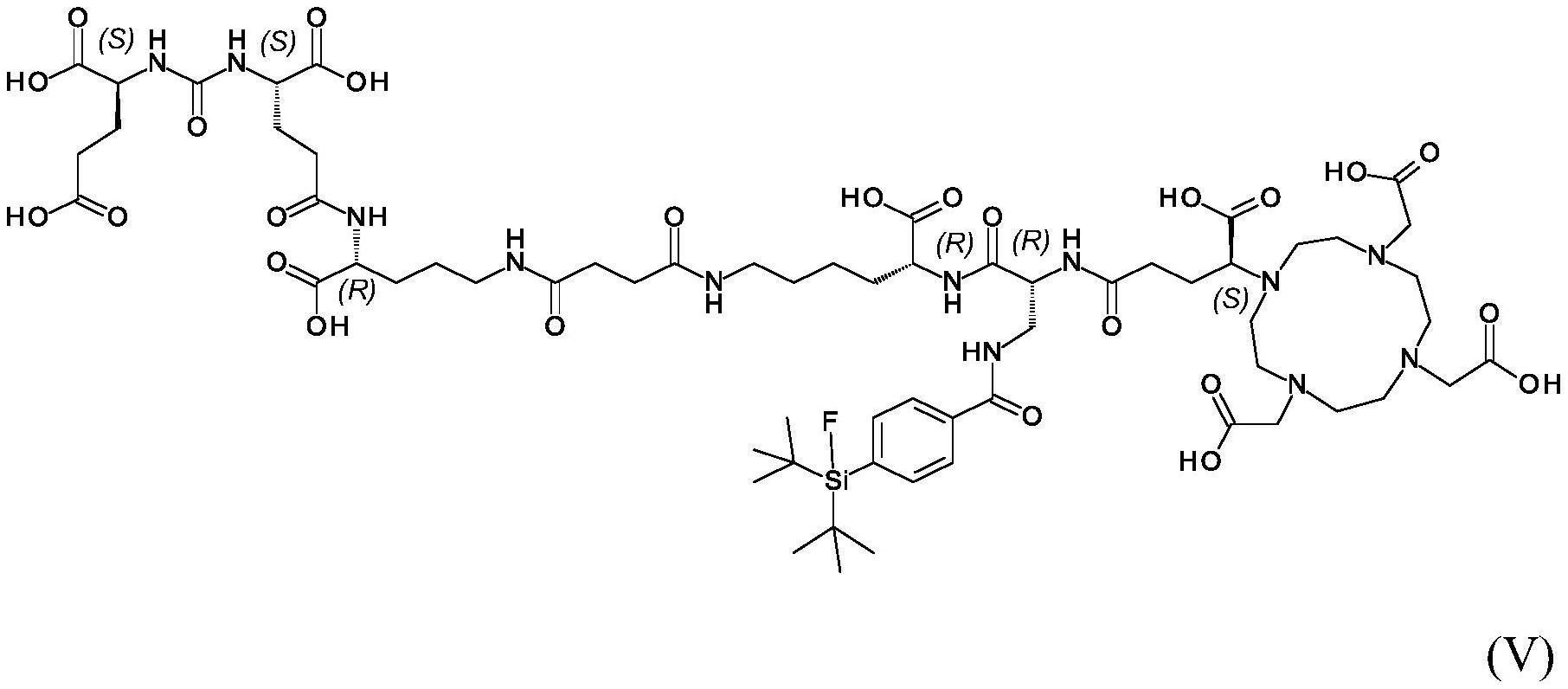
2.本发明涉及根据式(v)的化合物或其药学上可接受的盐:
[0003][0004]
其包含螯合的放射性阳离子或者其中f任选地为
18
f。本发明亦涉及在合成期间防止外消旋化的合成所述化合物的方法。
[0005]
在本说明书中,引用了包括专利申请和制造商手册在内的许多文件。尽管不认为这些文件的公开内容与本发明的专利性相关,但在此通过引用的方式整体并入。更具体而言,所有引用文件均通过引用的方式并入,其并入程度如同每个单独的文件均具体且独立地被指示通过引用的方式并入一般。
[0006]
前列腺癌
[0007]
近几十年,前列腺癌(pca)仍然是男性最常见的恶性疾病,发生率高,生存率低。由于在前列腺癌中的过表达,前列腺特异性膜抗原(psma)或谷氨酸羧肽酶ii(gcp ii)已证实了其作为开发用于pca的腔内放射疗法(endoradiotherapy)和成像的高灵敏放射性标记剂的优良靶标的合格性。前列腺特异性膜抗原为一种胞外水解酶,其催化中心包含两个锌(ii)离子与一个桥连氢氧基配体。虽然它在转移性和激素难治性前列腺癌中高度上调,但其在肾脏、唾液腺、小肠、脑中的生理表达以及在健康的前列腺组织中较低程度的生理表达亦有报道。在肠中,psma通过将蝶酰基聚-γ-谷氨酸盐转化成蝶酰谷氨酸盐(叶酸盐)来促进叶酸的吸收。在脑中,其将n-乙酰基-l天冬氨酰基-l-谷氨酸盐(naag)水解成n-乙酰基-l-天冬氨酸盐和谷氨酸盐。
[0008]
前列腺特异性膜抗原(psma)
[0009]
前列腺特异性膜抗原(psma)为一种ii型跨膜糖蛋白,其在前列腺癌上皮细胞上高度过表达。尽管其名为前列腺特异型膜抗原,但psma亦不同程度地在多种非前列腺癌的新血管系统中表达。在证实psma表达的最常见的非前列腺癌中包括乳腺癌、肺癌、结肠直肠癌和肾细胞癌。
[0010]
psma靶向分子的一般必要结构包括这样的结合单元,其包含与p1’谷氨酸酯部分连接的锌结合基团(例如尿素、次磷酸酯或氨基磷酸酯),这保证了对psma的高亲和力和特
异性,并且通常进一步连接到效应子官能团。所述效应子部分柔性更高且在某种程度上耐受结构修饰。入口通道容纳有对配体结合而言很重要的另外两个突出的结构特征。第一个为精氨酸补丁(patch),其为入口通道壁上带正电荷的区域并且是psma的p1位置上偏好带负电荷官能团的机理解释。这似乎是优选将带负电荷的残基并入配体-支架中的原因。据我们所知,目前尚未进行关于正电荷对psma配体的影响的深入分析。结合之后,精氨酸侧链的协同重新定位可导致s1疏水附囊的打开,这是第二个重要结构,其已被证明可容纳几种基于脲的抑制剂的碘苄基,从而有助于其对psma的高亲和力。
[0011]
zhang等发现了psma的远端结合位点,其可用于二齿结合模式(zhang等,journal of the american chemical society 132,12711-12716(2010))。所谓的芳烃结合位点是由arg463、arg511和trp541的侧链形成的简单结构基序,并且是gcpii入口盖的一部分。远端抑制剂部分对芳烃结合位点的接合可导致对psma的抑制剂亲和力因亲合性效应而大幅增加。尽管没有可用的结合模式的晶体结构分析,但出于以此方式与psma相互作用的意图开发了psmai&t。根据zhang等的必要特征为接头单元(在psma i&t的情况下为辛二酸),其促进gcpii入口盖的开放构象并从而使芳烃结合位点的可及性成为可能。进一步显示的是,所述接头的结构组成对肿瘤靶向性和生物学活性以及对成像对比度和药代动力学有重要影响(liu等,bioorganic&medicinal chemistry letters 21,7013-7016(2011)),所述特性对高成像质量和有效的靶向腔内放射治疗两者而言极其关键。
[0012]
目前在临床环境中使用两类psma靶向抑制剂。一方面存在具有用于放射性核素络合的螯合单元的示踪剂,例如psmai&t或相关化合物。另一方面存在小分子,其包含靶向单元和效应分子。
[0013]
用于选择性psma成像的最常用试剂是psma hbed-cc、psma-617和psma i&t,其主要用
68
ga(88.9%β
+
,e
β+,max
=1.89mev,t
1/2
=68min)标记。其中,
68
ga-psma-hbed-cc(亦称为
68
ga-psma-11)目前被认为是用于pca的pet成像的金标准。
[0014]
18
f标记
[0015]
近来,数个团体已致力于开发用于pca诊断的新型
18
f-标记的基于脲的抑制剂。与可获自商业分销的
68
ge/
68
ga放射性核素发生器(
68
ge;t
1/2
=270.8d)的放射性金属
68
ga相比,放射性同位素
18
f-氟化物(96.7%β
+
,e
β+,max
=634kev)需要现场回旋加速器用于其生产。尽管有此限制,但是就日常操作和成像质量而言,
18
f因其半衰期更长(t1/2=109.8min)及其正电子能量更低而呈现出显著优势。另外,有可能在回旋加速器中进行大规模生产,这将有益于更高的患者吞吐量并降低生产成本。
18
f-标记的基于脲的psma抑制剂
18
f-dcfpyl在原发性和转移性pca的检测中显示出有前景的结果(rowe等,molecular imaging and biology,1-9(2016)),并且在比较研究中优于
68
ga-psma-hbed-cc(dietlein等,molecular imaging and biology17,575-584(2015))。基于psma-617的结构,最近开发了
18
f-标记的类似物psma-1007,其显示了相当的肿瘤/器官比率(cardinale等,journal of nuclear medicine:核医学协会官方出版物58,425-431(2017);giesel等,european journal of nuclear medicine and molecular imaging 43,1929-1930(2016))。使用
68
ga-psma-hbed-cc的比较研究显示两种示踪物诊断准确度相似以及
18
f-psma-1007的尿清除率减少,使前列腺的更好评估成为可能(giesel等,european journal of nuclear medicine and molecular imaging 44,678-688(2017))。
[0016]
用于引入
18
f标记物的一种有吸引力的方法是使用硅氟化物受体(sifa)。硅氟化物受体例如在lindner等,bioconjugate chemistry 25,738-749(2014)中描述。为了保护硅-氟化物键,硅氟化物受体的使用引入了在硅原子周围的基团必然在空间上要求严格。这转而致使硅氟化物受体为高度疏水的。对于与靶分子的结合,具体而言,与靶分子即psma的结合,硅氟化物受体提供的疏水部分可用于建立放射性诊断化合物或放射性治疗化合物与疏水袋的相互作用的目的,其描述于zhang等,journal of the american chemical society 132,12711-12716(2010)中。然而,在结合之前,引入到分子中的较高程度的亲脂性就开发具有合适的体内生物分布即在非靶组织中有少量非特异性结合的放射性药物而言造成了严重的问题。
[0017]
未能解决疏水性问题
[0018]
尽管进行了许多尝试,但是用现有技术还没有圆满解决由硅氟化物受体引起的疏水性问题。
[0019]
为进一步解释,schirrmacher e.等(bioconjugate chem.2007,18,2085-2089)使用高效标记合成子对-(二叔丁基氟代甲硅烷基)苯甲醛([
18
f]sifa-a,硅氟化物受体的一个实例)合成了不同的
18
f-标记肽。sifa技术得到出人意料有效的同位素
19
f-18
f交换,并且无需应用hplc纯化便可以以介于225至680gbq/μmol(6081-18 378ci/mmol)之间的高比活性得到产量几乎定量的
18
f-合成子。最终将[
18
f]sifa-苯甲醛用于以高放射化学产率标记n-端氨基-氧代(n-ao)衍生肽ao-tyr3-octreotate(ao-tate)、环(fk(ao-n)rgd)和n-ao-peg
2-[d-tyr-gln-trp-ala-val-ala-his-thi-nle-nh2](ao-bzh3,铃蟾肽衍生物)。尽管如此,标记的肽仍为高度亲脂的(如可使用本文所述的条件从hplc保留时间得出),因此不适于在动物模型或人体中进一步评价。
[0020]
在c.等(bioconjugate chem.,2009,20(2),第317-321页)中,描述了蛋白质(大鼠血清白蛋白,rsa)的首次基于sifa的试剂盒样放射性氟化。作为标记试剂,通过简单同位素交换以40-60%放射化学产率(rcy)产生4-(二叔丁基[
18
f]氟代甲硅烷基)苯硫醇(si[
18
f]fa-sh)并在20-30min内以12%的总rcy将所述产物直接与马来酰亚胺衍生的血清白蛋白耦合。技术上简单的标记程序无需任何复杂的纯化程序,且为si-18
f化学成功应用于使用pet体内成像的简单实例。小鼠的时间-活性曲线和μpet影像显示大部分活性局限于肝中,从而证实标记试剂过于亲脂并将体内探针导向肝胆排泄和广泛的肝脏代谢。
[0021]
c.等(参见bioconjug chem.2010年12月15日;21(12):2289-96)随后尝试通过合成并评价新型sifa-octreotate类似物(sifa-tyr3-octreotate、sifa-asn(acnh-β-glc)-tyr3-octreotate和sifa-asn(acnh-β-glc)-peg-tyr3-octreotate)来克服sifa技术的主要缺点——所得放射性药物的高亲脂性。在这些化合物中,在肽和sifa部分之间引入亲水接头和药代动力学修饰物,即碳水化合物和peg接头加上碳水化合物。作为缀合物的亲脂性的量度(measure),确定了log p(ow)并发现sifa-asn(acnh-β-glc)-peg-tyr
3-octreotate为0.96以及sifa-asn(acnh-β-glc)-tyr
3-octreotate为1.23。这些结果显示,sifa部分的高亲脂性只能通过应用亲水性部分得到略微补偿。首次成像研究证实了过度的肝清除/肝摄取,并因此从未转入首次人体研究。
[0022]
bernard-gauthier等(biomed res int.2014;2014:454503)综述了已在文献中报
道的大量不同的sifa物质,范围从小辅基及其他低分子量化合物到标记的肽及最近的亲和体(affibody)分子。基于这些数据,至今仍未解决基于sifa的辅基的亲脂性问题;即尚未描述将sifa缀合肽的总亲脂性降至低于约-2.0的log d的方法。
[0023]
在lindner s.等(bioconjug chem.2014年4月16日;25(4):738-49)中,描述了合成作为特异性grp受体配体的聚乙二醇化铃蟾肽(pesin)衍生物和作为特异性αvβ3结合剂的rgd(精氨酸-甘氨酸-天冬氨酸的单字母代码)肽,并用硅-氟受体(sifa)部分标记。为了抵消sifa部分的高亲脂性,引入了各种亲水性结构修饰,得到降低的logd值。sifa-asn(acnh-β-glc)-pesin、sifa-ser(β-lac)-pesin、sifa-cya-pesin、sifa-lysme3-pesin、sifa-γ-羧基-d-glu-pesin、sifa-cya2-pesin、sifa-lysme3-γ-羧基-d-glu-pesin、sifa-(γ-羧基-d-glu)2-pesin、sifa-rgd、sifa-γ-羧基-d-glu-rgd、sifa-(γ-羧基-d-glu)2-rgd、sifa-lysme3-γ-羧基-d-glu-rgd。所有这些肽——已出于降低亲脂性的目的进行改良和衍生——均显示范围介于+2和-1.22之间的logd值。
[0024]
在niedermoser s.等(j nucl med.2015年7月;56(7):1100-5)中,将新开发的
18
f-sifa-修饰的tate衍生物和
18
f-sifalin-(sifa =硅氟化物受体)修饰的tate衍生物与用于荷生长激素抑制素受体肿瘤的高质量成像的现行临床金标准
68
ga-dotatate进行比较。出于这个目的,开发了
18
f-sifa-tate和两个相当复杂的类似物
18
f-sifa-glc-peg1-tate、
18
f-sifalin-glc-asp2-peg1-tate。所述试剂均未显示logd《-1.5。
[0025]
鉴于以上所述,可见本发明的根本技术问题在于提供这样的放射性诊断和放射性疗法,其含有硅氟化物受体并且同时以良好的体内特性为特征。
[0026]
如在下文中将明显的,本发明使用以高亲和力与前列腺特异性抗原(psma)结合的特异性缀合物作为靶标建立了原理验证(proof-of-principle)。因此,可见本发明的另一个根本技术问题在于提供用于医学指征(其为癌症,优选前列腺癌)的改进的放射性疗法和放射性诊断。
[0027]
pct/ep2018/070533公开了一组结合psma的化合物。本文公开的是来自在先申请的化合物的有利子集。本技术是发明人在提交pct/ep2018/070533时未理解的有利特征的选集(selection)。
[0028]
这些技术问题通过权利要求的主题来解决。因此,在一些方面,本发明涉及根据式(v)的化合物或其药学上可接受的盐:
[0029]
[0030]
其包含螯合的放射性阳离子或者其中f任选地为
18
f。
[0031]
公开的化合物可以是盐的形式。本发明亦涉及式(va)的化合物或其药学上可接受的盐:
[0032][0033]
其中;
[0034]
各x独立地为oh或o-;
[0035]
m为螯合的放射性阳离子或不存在;
[0036]
和f任选地为
18
f。
[0037]
另外,联合使用螯合剂和借由
18
f-氟化物在sifa上的同位素交换,亦在具有现场回旋加速器的中心或通过运输从回旋加速器中心获得
18
f-氟化物的中心得到“成对的”诊断示踪剂,,其可作为[
18
f][
nat
ion]示踪剂使用,而在无法获得
18
f-氟化物但可使用放射性同位素发生器(例如ge-68/ga-68发生器)的中心,可使用其对应形式,例如[
nat
f][
68
ga]示踪剂。
[0038]
重要的是,在两种情况下注入化学上一致的放射性药物,并因此预期在体内表现上无差异。然而目前因化学差异所致,由一个地点的患者组提供的
18
f-标记化合物的临床数据不能直接与由另一地点的另一个组提供的
68
ga-类似物的临床数据进行比较,而根据本发明的放射性药物和/或诊断剂可进行直接比较并因此将允许链接所述数据(例如来自用f-18操作的位于欧洲的一个中心的数据和用ga-68操作的位于印度的另一个中心的数据)。
[0039]
此外,在适当选择时,所述螯合物亦可用于使用治疗性同位素标记,例如发射β射线的同位素lu-177、y-90或发射α射线的同位素ac-225,从而允许将“成对的”示踪剂的概念扩展至桥接诊断剂([
18
f][
nat
lu]示踪剂)和治疗用放射性药物([
nat
f][
177
lu])。
[0040]
本发明化合物(尤其是psma靶向化合物)的另一个优点为当与其他psma靶向放射性药物(例如psmai&t)比较时,它们在小鼠肾脏出人意料较的低累积。不希望受特定理论的束缚,似乎是结构元件sifa与螯合剂的组合提供了在肾脏中累积的意想不到的减少。
[0041]
就亲脂性/亲水性而言,logp值(有时亦称为logd值)为本领域公认的量度。
[0042]
术语“亲脂性”是指溶解或吸收于脂质溶液中,或者吸附于脂质样表面或基质上的能力。其表示与水相比对脂质(字面含义),或者对有机液体或非极性液体,或者对偶极矩小的液体、溶液或表面的偏好。术语“疏水性”在本文中以等同的含义使用。形容词亲脂的和疏水的以与上述名词性实词相对应的含义使用。
[0043]
分子在两种不互溶的或基本不互溶的溶剂的界面处的质量流量由其亲脂性决定。
分子越亲脂,其在亲脂性有机相中越易溶。已采用在水和正辛醇之间观察到的分子的分配系数作为亲脂性的标准量度。物质a的分配系数p定义为比率p=[a]
正辛醇
/[a]
水
。通常报告的数字为logp值,为所述分配系数的对数。如果分子为可电离的,那么原则上在两相中都会存在多种不同的微量物质(所述分子的电离形式和非电离形式)。描述可电离物质的总亲脂性的量是分配系数d,定义为比率d=[所有微量物质的浓度之和]
正辛醇
/[所有微量物质的浓度之和]
水
。与logp类似,经常报告分配系数的对数logd。在上述logp的测定中,常常将例如磷酸缓冲盐水等缓冲体系用作水的替代。
[0044]
如果要评估和/或定量确定第一分子上的取代基的亲脂特性,可评估与所述取代基相对应的第二分子,其中所述第二个分子例如通过断开连接所述取代基与第一分子的剩余部分的键并将由此获得的一个或多个游离价与一个或多个氢连接而获得。
[0045]
或者,可确定取代基对分子的logp的贡献。取代基x对分子r-x的logp的贡献π
xx
定义为π
xx
=logp
r-x-logp
r-h
,其中r-h为未取代的母体化合物。
[0046]
大于1的p和d值以及大于0的logp、logd和π
xx
值指示亲脂/疏水特性,而小于1的p和d值以及小于0的logp、logd和π
xx
值指示相应分子或取代基的亲水特性。
[0047]
表征根据本发明的亲脂基团或整个分子的亲脂性的上述参数,可通过本领域已知的实验手段确定和/或通过计算方法预测(参见例如sangster,辛醇-水分配系数:基础与物理化学(octanol-water partition coefficients:fundamentals and physical chemistry),john wiley&sons,chichester.(1997)))。
[0048]
在一个优选的实施方案中,本发明化合物的logp值介于-5和-1.5之间。特别优选logp值介于-3.5和-2.0之间。
[0049]
在一个优选的实施方案中,所述螯合基团包含放射性的螯合阳离子。更优选螯合的放射性金属同位素。
[0050]
可被所述螯合基团螯合的阳离子的优选实例为
43
sc、
44
sc、
47
sc、
51
cr、
52m
mn、
58
co、
52
fe、
56
ni、
57
ni、
62
cu、
64
cu、
67
cu、
66
ga、
67
ga、
68
ga、
89
zr、
90
y、
89
y、《tc、
99m
tc、
97
ru、
105
rh、
109
pd、
111
ag,
110m
in、
111
in、
113m
in、
114m
in、
117m
sn、
121
sn、
127
te、
142
pr、
143
pr、
149
pm、
151
pm、
149
tb、
152
tb、
155
tb、
161
tb、
153
sm、
157
gd、
161
tb、
166
ho、
165
dy、
169
er、
169
yb、
175
yb、
172
tm、
177
lu、
186
re、
188
re、
191
pt、
197
hg、
198
au、
199
au、
212
pb、
203
pb、
211
at、
212
bi、
213
bi、
223
ra、
225
ac、
227
th的阳离子、包含
18
f的阳离子分子或例如
18
f-[alf]
2+
的阳离子;更优选
44
sc、
47
sc、
64
cu、
67
cu、
68
ga、
90
y、
111
in、
161
tb、
166
ho、
177
lu、
188
re、
212
pb、
212
bi、
213
bi、
225
ac和
227
th的阳离子或包含
18
f的阳离子分子。阳离子可选自lu-177、y-90或ac-225。
[0051]
在另一方面,本发明提供一种药物组合物,其包含如在上文中所公开的本发明的一种或多种化合物或者由如在上文中所公开的本发明的一种或多种化合物组成。
[0052]
在另一方面,本发明提供一种诊断组合物,其包含如在上文中所公开的本发明的一种或多种化合物或者由如在上文中所公开的本发明的一种或多种化合物组成。
[0053]
在另一方面,本发明提供一种治疗组合物,其包含如在上文中所公开的本发明的一种或多种化合物或者由如在上文中所公开的本发明的一种或多种化合物组成。
[0054]
所述药物组合物可进一步包含药学上可接受的载体、赋形剂和/或稀释剂。合适的药物载体、赋形剂和/或稀释剂的实例为本领域众所周知的且包括磷酸缓冲盐溶液、水、乳剂(例如油/水乳剂)、各种类型的润湿剂、无菌溶液等。包含所述载体的组合物可通过众所
周知的常规方法配制。可将这些药物组合物以合适的剂量施用给受试者。合适的组合物的施用可通过不同方式来实现,例如通过静脉内、腹膜内、皮下、肌内、局部、真皮内、鼻内或支气管内施用来实现。特别优选通过注射和/或递送到例如胰腺中的部位,或注射和/或递送到脑动脉中或直接注射和/或递递送到脑组织中来实施所述施用。亦可例如通过基因枪(biolistic)递送至外部或内部靶部位将所述组合物直接施用到靶部位,如胰腺或脑。剂量方案将由主治医师和临床因素来确定。如在医学领域众所周知的,用于任一位患者的剂量取决于许多因素,包括患者体型、体表面积、年龄、待施用的具体化合物、性别、给予时间和途径、一般健康状况和正在同时施用的其他药物。药学活性物质可以介于每剂量0.1ng和10mg/kg体重之间的量存在;然而,尤其是在考虑前述因素的情况下,可预想低于或高于此示例范围的剂量。
[0055]
在另一方面,本发明提供用于医学的如在上文中所公开的本发明的一种或多种化合物。
[0056]
在医学中优选的用途是在核医学中,例如核诊断成像(亦称为核分子成像),和/或与过表达(优选psma在患病组织上的过表达)相关的疾病的靶向放射疗法。
[0057]
在另一方面,本发明提供用于诊断和/或分期癌症(优选前列腺癌)的方法的如在上文中所定义的本发明的化合物。前列腺癌并非唯一表达psma的癌症。已知表现出psma表达的非前列腺癌症包括乳腺癌、肺癌、结肠直肠癌和肾细胞癌。因此,具有psma结合部分的本文所述的任何化合物均可用于诊断、成像或治疗具有psma表达的癌症。
[0058]
优选的适应症为癌症(例如但不限于高级别胶质瘤、肺癌,尤其是前列腺癌和转移性前列腺癌)的检测或分期,在患有中风险至高风险的原发性前列腺癌患者中转移性疾病的检测,以及在患有生化复发的前列腺癌的患者中甚至在低血清psa值的情况下转移性部位的检测。另一个优选的适应症为新血管形成的成像和可视化。
[0059]
就待接受治疗尤其是放射治疗的医学适应症而言,癌症为优选的适应症。前列腺癌为特别优选的适应症。
[0060]
在另一方面,本发明提供用于诊断和/或分期癌症(优选前列腺癌)的方法中的如在上文中所定义的本发明的化合物。
[0061]
本公开还涉及以下项目。
[0062]
根据式(v)的化合物或其药学上可接受的盐:
[0063]
[0064]
其包含螯合的放射性阳离子或其中f任选地为
18
f。
[0065]
所述化合物可包含选自sc、cu、ga、y、in、tb、ho、lu、re、pb、bi、ac、er和th的阳离子的螯合的阳离子。所述螯合的阳离子可以是放射性的。所述螯合的放射性阳离子可以是镓、铒、铜、钪、镥或钇的任何放射性同位素。
[0066]
式(va)的化合物或其药学上可接受的盐:
[0067][0068]
其中;
[0069]
每个x独立地为oh或o-;
[0070]
m为螯合的放射性阳离子或不存在;
[0071]
并且f任选地为
18
f。
[0072]
在式(va)的化合物中,x可以是oh。x可以是o-。当m存在时,一个或多个x基团可与m螯合。
[0073]
在式(va)的化合物中,m可以是螯合的放射性阳离子。m可不存在。m可以是与一个或多个x基团螯合的放射性阳离子。m可以是与一个或多个n原子螯合的放射性阳离子。m可以是与一个或多个n原子或者一个或多个x基团螯合的放射性阳离子。m可以是与一个或多个n原子和一个或多个x基团螯合的放射性阳离子。
[0074]
在式(v)或(va)的化合物中,f可以是
18
f。f可以是
19
f。
[0075]
在式(v)或(va)的化合物中,所述螯合的放射性阳离子可选自
43
sc、
44
sc、
47
sc、
51
cr、
52m
mn、
58
co、
52
fe、
56
ni、
57
ni、
62
cu、
64
cu、
67
cu、
66
ga、
67
ga、
68
ga、
89
zr、
90
y、
89
y、《tc、
99m
tc、
97
ru、
105
rh、
109
pd、
111
ag,
110m
in、
111
in、
113m
in、
114m
in、
117m
sn、
121
sn、
127
te、
142
pr、
143
pr、
149
pm、
151
pm、
149
tb、
152
tb、
155
tb、
161
tb、
153
sm、
157
gd、
161
tb、
166
ho、
165
dy、
169
er、
169
yb、
175
yb、
172
tm、
177
lu、
186
re、
188
re、
191
pt、
197
hg、
198
au、
199
au、
212
pb、
203
pb、
211
at、
212
bi、
213
bi、
223
ra、
225
ac、
227
th的阳离子、包含
18
f的阳离子分子或例如
18
f-[alf]
2+
的阳离子;更优选
44
sc、
47
sc、
64
cu、
67
cu、
68
ga、
90
y、
111
in、
161
tb、
166
ho、
177
lu、
188
re、
212
pb、
212
bi、
213
bi、
225
ac和
227
th的阳离子或包含
18
f的阳离子分子。所述螯合的放射性阳离子可选自sc、cu、ga、y、in、tb、ho、lu、re、pb、bi、ac、er和th的阳离子。所述螯合的放射性阳离子可以是ga。所述螯合的放射性阳离子可以是lu-177、y-90或ac-225。
[0076]
在式(va)的化合物中,m可选自
43
sc、
44
sc、
47
sc、
51
cr、
52m
mn、
58
co、
52
fe、
56
ni、
57
ni、
62
cu、
64
cu、
67
cu、
66
ga、
67
ga、
68
ga、
89
zr、
90
y、
89
y、《tc、
99m
tc、
97
ru、
105
rh、
109
pd、
111
ag,
110m
in、
111
in
、
113m
in、
114m
in、
117m
sn、
121
sn、
127
te、
142
pr、
143
pr、
149
pm、
151
pm、
149
tb、
152
tb、
155
tb、
161
tb、
153
sm、
157
gd、
161
tb、
166
ho、
165
dy、
169
er、
169
yb、
175
yb、
172
tm、
177
lu、
186
re、
188
re、
191
pt、
197
hg、
198
au、
199
au、
212
pb、
203
pb、
211
at、
212
bi、
213
bi、
223
ra、
225
ac、
227
th的阳离子、包含
18
f的阳离子分子或例如
18
f-[alf]
2+
的阳离子;更优选
44
sc、
47
sc、
64
cu、
67
cu、
68
ga、
90
y、
111
in、
161
tb、
166
ho、
177
lu、
188
re、
212
pb、
212
bi、
213
bi、
225
ac和
227
th的阳离子或包含
18
f的阳离子分子。m可选自sc、cu、ga、y、in、tb、ho、lu、re、pb、bi、ac、er和th的阳离子。m可以是ga。m可以是lu-177、y-90或ac-225。
[0077]
有利的是合成异构体尽可能少的化合物。尽管可对异构体进行分离,但是在体内仅使用单个异构体因此错误的异构体只能被丢弃并不使用。因此,理想地要避免使任何确定的手性中心的外消旋化或反转减至最少的条件。
[0078]
亦描述了一种产生所述化合物的方法,包括以下步骤:
[0079]
a)使式(i)的化合物:
[0080][0081]
与式(ii)的化合物:
[0082][0083]
反应形成式(iii)的化合物:
[0084][0085]
其中pg1为tbu,pg2为fmoc和pg3为dde;
[0086]
以及所述反应条件包括碱的使用,其中所述碱为2,4,6-三甲基吡啶或2,6-二甲基
吡啶;
[0087]
b)使式(iii)的化合物在适于形成式(iv)的化合物的条件下反应:
[0088][0089]
和
[0090]
c)使式(iv)的化合物在适于形成化合物(v)的条件下反应:
[0091][0092]
相应的方法包括其中化合物(ii)通过与2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲四氟硼酸酯(tbtu)、1-羟基-7-氮杂苯并三唑(hoat)和2,4,6-三甲基吡啶的反应预活化,然后与化合物(i)反应。所述预活化进行5分钟或更少时间。
[0093]
与其他含氮碱(例如dipea)相比,使用2,4,6-三甲基吡啶或2,6-二甲基吡啶作为碱连同较短的活化时间,有助于将活化的手性化合物(ii)的外消旋化减至最少。空间位阻较大的碱不会提取酸的手性中心上的酸性质子,从而在与胺耦联之前减少外消旋化。
[0094]
本文公开了根据式(vi)的化合物或其药学上可接受的盐。
[0095][0096]
当显示螯合的金属时,与之螯合的酸基团仅代表性地显示为coo-,等同的第四个酸亦可能被部分螯合并且因此可能不是字面上的cooh。
[0097]
本文公开了根据式(vii)的化合物或其药学上可接受的盐。
[0098][0099]
本文公开了一种药物组合物或诊断组合物,其包含式(v)或(vi)的化合物。所述缀合物、化合物或组合物可用作癌症诊断剂或成像剂。
[0100]
公开了一种对癌症进行成像和/或诊断的方法,其包括向有需要的患者施用根据式(v)或(vi)的缀合物、化合物或组合物。
[0101]
公开了用于癌症的治疗中的根据式(v)或(vi)的缀合物、化合物或组合物。
[0102]
公开了用于新血管形成/血管形成的诊断、成像或预防的根据式(v)或(vi)的缀合物、化合物或组合物。
[0103]
公开了用作癌症诊断剂或成像剂或者用于癌症的治疗的根据式(v)或(vi)的缀合物、化合物或组合物,其中所述癌症为前列腺癌、乳腺癌、肺癌、结肠直肠癌或肾细胞癌。
附图说明:
[0104]
图1:在originpro 2016g中测定9种参考物质的示例性相关性。
[0105]
图2a:[19f][natga]-rhpsma7-rac([19f][natga]d/l-dap-r/s-dotaga-rhpsma-7-rac),(批次10,在tum的核医学系用于生产([18f][natga]d/l-dap-r/s-dotaga-rhpsma-7的前体)的质量控制。hplc条件:溶剂a:h20+0.1% tfa;溶剂b:mecn+0.1% tfa。梯度:25-35% b 0-40min,95-95% b 40-45min,35-35% b 45-50min;流速:1ml/min,柱:nucleosil100-5c18,125x 4.6mm,样品:1mm(dmso),10μl。
[0106]
图2b:峰归属:d-dap-r-dotaga-rhpsma-7.1;与对映异构体纯的d-dap-r-dotaga-rhpsma-7.1共注入的来自图2a的rhpsm7-rac。hplc条件:溶剂a:h20+0.1% tfa;溶剂b:
mecn+0.1% tfa。梯度:25-35% b 0-40min,95-95% b 40-45min,35-35% b 45-50min;流速:1ml/min,柱:nucleosil 100-5c18,125x 4.6mm,样品:1mm(dmso),10μl。
[0107]
图2c:d-dap-r-dotaga-rhpsma-7.1的hplc图谱;hplc条件:溶剂a:h20+0.1% tfa;溶剂b:mecn+0.1% tfa。梯度:25-35% b0-40min,95-95% b 40-45min,35-35% b 45-50min;流速:1ml/min,柱:nucleosil 100-5c18,125x 4.6mm,样品:1mm(dmso),10μl。
[0108]
图3a:峰归属:l-dap-r-dotaga-rhpsma-7.2;与对映异构体纯的l-dap-r-dotaga-rhpsma-7.2共注入的来自图2a的rhpsm7-rac。hplc条件:溶剂a:h20+0.1% tfa;溶剂b:mecn+0.1% tfa。梯度:25-35% b 0-40min,95-95% b 40-45min,35-35% b 45-50min;流速:1ml/min,柱:nucleosil 100-5c18,125x 4.6mm,样品:1mm(dmso),10μl。
[0109]
图3b:l-dap-r-dotaga-rhpsma-7.2的hplc图谱;hplc条件:溶剂a:h20+0.1% tfa;溶剂b:mecn+0.1% tfa。梯度:25-35% b0-40min,95-95% b 40-45min,35-35% b 45-50min;流速:1ml/min,柱:nucleosil 100-5c18,125x 4.6mm,样品:1mm(dmso),10μl。
[0110]
图4a:峰归属:d-dap-s-dotaga-rhpsma-7.3;与对映异构体纯的d-dap-s-dotaga-rhpsma-7.3共注入的来自图2a的rhpsm7-rac。hplc条件:溶剂a:h20+0.1% tfa;溶剂b:mecn+0.1% tfa。梯度:25-35% b 0-40min,95-95% b 40-45min,35-35% b 45-50min;流速:1ml/min,柱:nucleosil 100-5c18,125x 4.6mm,样品:1mm(dmso),10μl。
[0111]
图4b:d-dap-d-dotaga-rhpsma-7.3的hplc图谱;hplc条件:溶剂a:h20+0.1% tfa;溶剂b:mecn+0.1% tfa。梯度:25-35% b0-40min,95-95% b 40-45min,35-35% b 45-50min;流速:1ml/min,柱:nucleosil 100-5c18,125x 4.6mm,样品:1mm(dmso),10μl。
[0112]
图5a:峰归属:l-dap-s-dotaga-rhpsma-7.4;与对映异构体纯的d-dap-s-dotaga-rhpsma-7.3共注入的来自图2a的rhpsm7-rac。hplc条件:溶剂a:h20+0.1% tfa;溶剂b:mecn+0.1% tfa。梯度:25-35% b 0-40min,95-95% b 40-45min,35-35% b 45-50min;流速:1ml/min,柱:nucleosil 100-5c18,125x 4.6mm,样品:1mm(dmso),10μl。
[0113]
图5b:l-dap-d-dotaga-rhpsma-7.4的hplc图谱;hplc条件:溶剂a:h20+0.1% tfa;溶剂b:mecn+0.1% tfa。梯度:25-35% b0-40min,95-95% b 40-45min,35-35% b 45-50min;流速:1ml/min,柱:nucleosil 100-5c18,125x 4.6mm,样品:1mm(dmso),10μl。
[0114]
图6a:rhpsma7.1和rhpsma7.2与psma的结合亲和力(ic50[nm])。使用lncap细胞(150000个细胞/孔)和((4-[125i]碘代苯甲酰基)kue([125i]ib-kue;c=0.2nm)作为放射性配体(1h,4℃,hbss+1% bsa)来确定亲和力。数据表示为平均值
±
sd(n=3,在3个不同实验中)。
[0115]
图6b:rhpsma7异构体与psma的结合亲和力[nm]的确定。四列中的每一列显示对rhpsam7.1(左)至rhpsma7.4(右)的个体亲和力测量值。条件如在对图6a的说明中所述。
[0116]
图7:图6a和6b中显示的个体ic50[nm]测量值的描绘。删除了rhpsma7.1的5号值。条件如在对图6a的说明中所述。
[0117]
图8:个体内化测量值[[
125
i]ib-kue]的%]的描绘。作为参照配体([
125
i]i-ba)kue(c=0.2nm)的%的1小时处的内化活性(c=0.5nm),在lncap细胞(37℃,dmem f12+5% bsa,125000个细胞/孔)上确定。针对非特异性结合(10μmol pmpa)校正数据并将其表示为平均值
±
sd(n=3)。
[0118]
图9:rhpsma异构体的logp’s的个体测量值的描绘。
[0119]
图10:经
18
f-标记的rhpsma示踪剂在注射后1h在荷lncap肿瘤的scid小鼠中的生物分布(以%id/g计)。数据表示为平均值
±
sd(对于rhpsma7.1 n=4,对于rhpsma7.2 n=5,对于rhpsma7.3 n=4,对于rhpsma7.4 n=5和对于rhpsma7-rac n=3)。
[0120]
图11:与pmpa(8mg/kg)共注射的
18
f-rhpsma在注射后1h在荷lncap肿瘤的scid小鼠中的生物分布[%id/g]。数据表示为平均值
±
sd(n=3)。
[0121]
图12:通过酰胺键的代谢裂解生成的可能的物质。il:裂解形成具有增加的亲脂性的物质;df:脱氟;nd:不可检测,因为并非放射性的。
[0122]
图13:左:重叠峰1(rhpsma7.2)和2(rhpsma7.3)的图解分析;右:通过systat peakfit软件对峰形去卷积并积分;上:拟合的实验数据,下:去卷积的单峰。
[0123]
图14a:对相对变化(以注入外消旋混合物的变化%)进行定量以评价峰4(rhpsma7.1)的经典积分(通过hplc程序)和去卷积(通过
‘
peakfit’)的重现性。对于该峰,两种方法显示相似的性能。
[0124]
图14b:对相对变化(以注入外消旋混合物的变化%)进行定量以评价峰3(rhpsma7.4)的经典积分(通过hplc程序)和去卷积(通过
‘
peakfit’)的重现性。对于该峰,两种方法显示相似的性能。
[0125]
图15:各rhpsam7.1-7.4异构体在血液、肝脏、肾脏、肿瘤和尿液中相对于其在注射溶液([
18
f][
nat
ga]rhpsma7-rac)中的比例的百分比变化。数据表示为平均值
±
sd(n=4;亦参见图16)。
[0126]
图16:用于每个样品和实验的每个rhpsam7.1-7.4异构体在血液、肝脏、肾脏、肿瘤和尿液中相对于其在注射溶液([
18
f][
nat
ga]rhpsma7-rac)中的比例的百分比变化。使用systat peakfit进行分析。
[0127]
图17:左:载有肝脏样品的tlc板的tlc扫描图谱(2018年7月30日,总cts:142cts)。因其较差的统计学和有限的有效性,删除了cts《200的数据集。右:载有肝脏样品的tlc板的磷光图像(phophoimage):移动示踪剂的长拖尾。
[0128]
图18:左:载有质量控制样品的tlc板的tlc扫描图谱(2018年8月1日,总cts:384)。右:载有尿液、肾脏、肝脏、肿瘤、血液和qk样品的tlc板的示例性磷光图像。
[0129]
图19:在示踪剂的临床应用之前,作为核医学系质量控制的一部分对[f-18]rhpsma7-rac进行的放射性tlc分析(2018年7月30日)。注意,即便在配制缓冲液中(因此在不含蛋白质的条件下)亦观察到所述示踪剂的拖尾。
[0130]
图20:通过放射性-tlc对尿样中的游离[f-18]氟化物和
‘
完整的’[f-18]rhpsma7-rac的定量(2018年7月30日)。
[0131]
图21:左:从4只注射了各[f-18]rhpsam-7.x示踪剂的正常小鼠收集并合并的尿液的放射性hplc分析。
[0132]
右:掺入各[f-18]rhpsam-7.x示踪剂持续1h(7.1.、7.2.)、0.5h(7.3.)和2h(7.4.)时间段的
‘
冷’尿液的放射性hplc分析。
[0133]
hplc条件:溶剂a:h20+0.1% tfa;溶剂b:mecn+0.1% tfa;梯度:5%等度0-3min,25-35% b 3-43min,95-95% b 43-48min;流速:1ml/min,柱:nucleosil 100-5c18,125x 4.6mm。
[0134]
图22:尿液中的放射性物质通过滤筒固定和tlc的分离。
[0135]
上:在小鼠中注射[f-18]rhpsma7.3 30min后的尿液的放射性hplc分析在1.6min显示小比例示踪剂且在约34.5min显示完整示踪剂。
[0136]
下(左):在腹腔注射(p.i.)[f-18]rhpsma7.3 30min后,将小鼠的尿液稀释并进行strata-x滤筒固定。使用mecn/水(60/40v/v+1% tfa)洗涤和洗脱滤筒;仅检测到完整示踪剂。
[0137]
下(右):通过tlc分析来自滤筒固定的穿透物(breakthrough)(非保留组分)和最终用mecn/水从滤筒上洗脱的级分(下,右)。在滤筒的洗脱物中发现了96.1%[f-18]rhpsma7.3和仅3.9%[f-18]氟化物,而在滤筒的穿透物中发现了相反的比率(3.4%[f-18]rhpsma7.3和仅96.6%[f-18]氟化物)。
[0138]
图23:向小鼠的新鲜和非放射性尿液中加入[f-18]rhpsma7.3,接着加入0.5μmol冷的f-19-氟化物;孵育2h。
[0139]
放射性被完全(98.5%)转化至代表[f-18]氟化物的极亲水级分(1.6min处的峰)。注意:随后将1.6min处的峰固定在qma滤筒上并使用nacl(1m)洗脱(=氟化物)。
[0140]
图24:如通过suv最大值所展示的18f-rhpsma-7(左)和18f-rhpsma-7.3(右)在肿瘤病灶中的临床生物分布和摄取。数据表示为平均值
±
sd。
[0141]
图25:通过suv平均值所展示的
18
f-rhpsma-7(左)和
18
f-rhpsma-7.3(右)在肿瘤病灶中的临床生物分布和摄取。数据表示为平均值
±
sd。
[0142]
图26:通过suv最大值与背景的比率所展示的
18
f-rhpsma-7(左)和
18
f-rhpsma-7.3(右)在肿瘤病灶中的临床生物分布和摄取。数据表示为平均值
±
sd。
[0143]
图27:通过suv平均值与背景的比率所展示的
18
f-rhpsma-7(左)和
18
f-rhpsma-7.3(右)在肿瘤病灶中的临床生物分布和摄取。数据表示为平均值
±
sd。
[0144]
图28:
18
f-rhpsma-7.3pet成像的两个临床案例实例。
[0145]
实施例说明本发明。
[0146]
实施例1:材料和方法
[0147]
fmoc-(9-芴甲氧羰基-)和所有其他受保护的氨基酸类似物购自bachem(bubendorf,瑞士)或iris biotech(marktredwitz,德国)。三苯甲基氯聚乙烯(tcp)树脂获自pepchem(t
ü
bingen,德国)。chematech(dijon,法国)交付了螯合剂dotaga酐、(r)-dota-ga(tbu)4和(s)-dota-ga(tbu)4。所有必需溶剂及其他有机试剂均购自alfa aesar(karlsruhe,德国)、sigma-aldrich(munich,德国)或vwr(darmstadt,德国)。使用intelli-mixer注射器振荡器(neolab,heidelberg,德国)通过手动操作进行肽的固相合成。使用各自配备有spd-20auv/vis检测器(220nm、254nm)的shimadzu梯度系统(shimadzu deutschland gmbh,neufahrn,德国)进行分析型和制备型反相高压色谱(rp-hplc)。使用nucleosil 100c18(125
×
4.6mm,5μm粒径)柱(cs gmbh,langerwehe,德国)以1ml/min的流速进行分析测定。具体梯度和相应保留时间tr二者均在文本中引用。使用multospher 100rp 18(250
×
10mm,5μm粒径)柱(cs gmbh,langerwehe,德国)以5ml/min的恒定流速完成制备型hplc纯化。使用nucleosil 100c18(5μm,125
×
4.0mm)柱(cs gmbh,langerwehe,德国)进行分析型和制备型放射性rp-hplc。用于所有hplc操作的洗脱液均为水(溶剂a)和乙腈(溶剂b),二者均含0.1%三氟乙酸。在expression
l cms质谱仪(advion ltd.,harlow,英国)上获得用于表征物质的电喷雾电离质谱。用bruker avhd-300或avhd-400光谱仪在300k
记录nmr谱。使用seveneasy ph计(mettler toledo,gieβen,德国)测定ph值。
[0148]
合成方案
[0149]
1)遵循fmoc-策略的固相肽合成
[0150]
tcp-树脂负载(gp1)
[0151]
通过在室温下搅拌tcp-树脂(1.95mmol/g)和fmoc-aa-oh(1.5当量)于含dipea的无水dcm(4.5当量)中的溶液2h来进行用fmoc-保护氨基酸(aa)负载三苯甲基氯聚乙烯(tcp)树脂。通过加入甲醇(2ml/g树脂)达15min来封闭剩余的三苯甲基氯。随后将树脂过滤,用dcm(2
×
5ml/g树脂)、dmf(2
×
5ml/g树脂)、甲醇(5ml/g树脂)洗涤并在真空中干燥。fmoc-aa-oh的最终负载量l通过下式来确定:
[0152][0153]
m2=负载的树脂的质量[g]
[0154]
m1=未负载的树脂的质量[g]
[0155]mw
=aa的分子量[g/mol]
[0156]mhcl
=hcl的分子量[g/mol]
[0157]
树脂上的酰胺键形成(gp2)
[0158]
对于构建单元与树脂结合肽的缀合,使用tbtu和hobt的混合物以便用dipea或2,4,6-三甲基吡啶(作为碱在dmf中的溶液(10ml/g树脂))预活化5min。在合成方案中给出了各缀合步骤的确切的化学计量和反应时间。反应之后,用dmf(6
×
5ml/g树脂)洗涤树脂。
[0159]
树脂上的fmoc-脱保护(gp3)
[0160]
使用20%哌啶在dmf中的溶液(v/v,8ml/g树脂)将树脂结合的fmoc-肽处理5min并随后处理15min。然后,用dmf(8
×
5ml/g树脂)彻底洗涤树脂。
[0161]
树脂上的dde-脱保护(gp4)
[0162]
将dde-保护的肽(1.0当量)溶于2%一水合肼在dmf中的溶液(v/v,5ml/g树脂)中并振荡20min(gp4a)。在存在fmoc基团的情况下,通过在室温下加入咪唑(0.92g/g树脂)、盐酸羟胺(1.26g/g树脂)于nmp(5.0ml)和dmf(1.0ml)中的溶液保持3h来进行dde-脱保护(gp4b)。脱保护之后,用dmf(8
×
5ml/g树脂)洗涤树脂。
[0163]
从树脂中裂解肽同时对酸不稳定性保护基脱保护(gp5)
[0164]
将完全保护的树脂结合肽溶于tfa/tips/水的混合物(v/v/v;95/2.5/2.5)中并振荡30min。滤出溶液并以相同方式将树脂再处理30min。将两次滤液合并,再搅拌5h并在氮气流下浓缩。将残留物溶于叔丁醇和水的混合物中并随后冷冻干燥,得到粗肽。
[0165]
nat
ga-络合(gp6)
[0166]
对于
nat
ga-络合,将肽(1.0当量)溶于tbuoh/h2o的3:1(v/v)混合物中并加入ga(no3)3(3.5当量)的水溶液。将所得混合物于75℃加热30min之后,通过rp-hplc纯化肽。
[0167]
2)psma结合基序的合成
[0168]
glu-尿素-glu((tbuo)eue(otbu)2)
[0169][0170]
根据先前公布的用于tbu-保护的glu-尿素-lys(euk)的程序(方案1)来合成tbu-保护的glu-尿素-glu结合基序(eue)。
[0171]
(1h-咪唑-1-羰基)-l-谷氨酸二叔丁酯(i)
[0172]
将含2.0g(7.71mmol,1.0当量)l-谷氨酸-i二叔丁酯
·
hcl的dcm溶液在冰上冷却30min,然后用2.69ml tea(19.28mmol,2.5当量)和3.3mg(0.3mmol,0.04当量)dmap处理。在另外搅拌5min之后,在30min的时间段内缓慢加入溶于dcm中的1.38g(8.84mmol,1.1当量)1,1
’‑
羰基二咪唑(cdi)。将反应混合物进一步搅拌过夜并使之升温到室温。使用8ml饱和nahco3终止反应,同时进行水(2
×
)和盐水(2
×
)的洗涤步骤并用na2so4干燥。在真空中去除剩余溶剂,粗产物(s)-2-(1h-咪唑-1-甲酰胺基)戊二酸二叔丁酯(i)无需进一步纯化便可使用。
[0173]
(((s)-1,5-二-叔丁氧基-1,5-二氧代戊烷-2-基)氨基甲酰基)-l-谷氨酸5-苄1-(叔丁酯)(ii)
[0174]
将2.72g(7.71mmol,1.0当量)的粗产物(s)
‑‑
2-(1h-咪唑-1-羧酰胺基)戊二酸二叔丁酯(i)溶于1,2-二氯乙烷(dce)中并在冰上冷却30min。向该溶液中加入2.15ml(15.42mmol,2.0当量)tea和2.54g(7.71mmol,1.0当量)h-l-glu(obzl)-otbu
·
hcl并将所述溶液于40℃搅拌过夜。蒸发剩余溶剂,并使用硅胶快速色谱法用含乙酸乙酯/己烷/tea(500:500:0.8;v/v/v)的洗脱混合物纯化粗产物。去除溶剂之后,得到呈无色油状的(((s)-1,5-二-叔丁氧基-1,5-二氧代戊烷-2-基)氨基甲酰基)-l-谷氨酸5-苄1-(叔丁酯)(ii)。(tbuo)eue(otbu)2(iii)
[0175]
对于合成(tbuo)eue(otbu)2,将3.17g(5.47mmol,1.0当量)的(((s)-1,5-二-叔丁氧基-1,5-二氧代戊烷-2-基)氨基甲酰基)-l-谷氨酸5-苄1-(叔丁酯)(ii)溶于75ml etoh中并向该溶液中加入0.34g(0.57mmol,0.1当量)活性炭负载钯(10%)。先用h2吹扫含有反应混合物的烧瓶并在低h2压力(气囊)下将所述溶液于室温搅拌过夜。通过硅藻土纯化粗产物并在真空中蒸发溶剂。得到呈吸湿性固体的产物(iii)(84%)。hplc
[0176]
(15min内10%-90% b):tr=11.3min。计算的单同位素质量(c
23h49
n2o9):488.3;实测值:m/z=489.4[m+h]
+
,516.4[m+na]
+
。
[0177][0178]
方案1(tbuo)eue(otbu)2的合成:a)dci、tea、dmap(dcm);b)h-l-glu(obzl)-otbu
·
hcl、tea(dce);c)pd/c(10%)、h2(etoh)。
[0179]
3)硅-氟化物受体的合成
[0180]
4-(二-叔丁基氟代甲硅烷基)苯甲酸(sifa-ba)
[0181][0182]
根据先前公布的程序(方案2)来合成sifa-ba。所有反应均在干燥反应容器中使用真空气体歧管在氩气下进行。
[0183]
((4-溴代苄基)氧代)(叔丁基)二甲基硅烷(i)
[0184]
向4-溴苄基醇(4.68g,25.0mmol,1.0当量)于无水dmf(70ml)中的搅拌溶液中加入咪唑(2.04g,30.0mmol,1.2当量)和tbdmscl(4.52g,30.0mmol,1.2当量),并将所得混合物于室温搅拌16h。然后将所述混合物倒入冰冷的h2o(250ml)中并用et2o(5
×
50ml)萃取。将合并的有机级分用饱和nahco3水溶液(2
×
100ml)和盐水(100ml)洗涤、干燥、过滤并在真空中浓缩得到粗产物,通过快速柱色谱法(二氧化硅,5% etoac/汽油)纯化所述粗产物得到呈无色油状的i(7.18g,95%)。1h nmr(400mhz,cdcl3):δ[ppm]=0.10(6h,s,sime2t-bu),0.95(9h,s,sime2tbu),4.69(2h,s,ch2osi),7.21(2h,d),7.46(2h,d)。hplc(15min内50-100%b):tr=15min。
[0185]
二-叔丁基{4-[(叔丁基二甲基甲硅烷基氧代)甲基]苯基}氟硅烷(ii)
[0186]
在-78℃磁力搅拌下,向((4-溴代苄基)氧代)(叔丁基)二甲基硅烷(i)(1.56g,5.18mmol,1.0当量)于干thf(15ml)中的溶液中加入tbuli于戊烷中的溶液(7.29ml,1.7mol/l,12.4mmol 2.4当量)。将反应混合物于-78℃搅拌30min之后,在30min的时间段内将所得混悬液逐滴加至二-叔丁基二氟代硅烷(1.12g,6.23mmol,1.2当量)于干thf(10ml)中的冷却(-78℃)溶液中。在12h的时间段内使反应混合物温热至室温,然后用饱和nacl水溶液(100ml)进行水合。分离有机层并用乙醚(3
×
50ml)萃取水层。用硫酸镁干燥合并的有机层并过滤。将滤液在真空中浓缩,得到呈浅黄色油状的ii(1.88g,95%)。无需进一步纯化便可将其用于后续反应。nmr谱与文献[2]中报道的数据一致。hplc(20min内50-100% b):tr=19min。4-(二-叔丁基氟代硅烷基)苄醇(iii)
[0187]
向ii(1.88g,4.92mmol,1.0当量)于甲醇(50ml)中的混悬液中加入催化量的浓hcl水溶液(0.5ml)。将反应混合物于室温搅拌18h,然后在减压下去除溶剂和挥发物。用乙醚(40ml)将残留物再溶解并用饱和nahco3水溶液洗涤所述溶液。用乙醚(3
×
50ml)萃取水层。用硫酸镁将合并的有机层干燥并过滤。将滤液在真空中浓缩,得到凝固的呈浅黄色油状的iii(1.29g,98%)。无需进一步纯化便可使用所述产物。nmr谱与文献[2]中报道的数据一致。hplc(15min内50-100% b):tr=8.2min。
[0188]
4-(二-叔丁基氟代甲硅烷基)苯甲醛(iv)
[0189]
将醇iii(1.37g,5.10mmol,1.0当量)于干二氯甲烷(20ml)中的溶液逐滴加入倒氯铬酸吡啶盐(2.75g,12.8mmol,2.5当量)于干二氯甲烷(60ml)中的搅拌的冰冷混悬液中。将反应混合物于0℃搅拌30min且于室温搅拌2.5h之后,加入无水乙醚(40ml)并从黑色胶状物料中倾出上清液。用乙醚充分洗涤不溶性物料,并使合并的有机相通过短硅胶垫(每g粗产物10cm)以进行过滤。在真空中去除溶剂,得到呈浅黄色油状的醛iv(1.31g,96%)。nmr谱与文献[2]中报道的数据一致。hplc(15min内50-100% b):tr=10.5min。
[0190]
4-(二-叔丁基氟代甲硅烷基)苯甲酸(v)
[0191]
在室温下,向iv(1.31g,4.92mmol,1.0当量)、叔丁醇(30ml)、二氯甲烷(3.3ml)和ph 4.0-4.5的1.25m nah2po4·
h2o缓冲液(20ml)的混合物中加入1m kmno4水溶液(30ml)。将所述混合物搅拌25min之后,使之冷却至5℃,随即加入过量kmno4(0.78g,4.92mmol,1.0当量)。然后通过加入饱和na2so3水溶液(50ml)来淬灭反应。在加入2m hcl水溶液之后,所有mno2均溶解。用乙醚(3
×
100ml)萃取所得溶液。将合并的有机层用饱和nahco3水溶液洗涤、用mgso4干燥、过滤并在减压下浓缩,得到白色固体,通过从et2o/正己烷(1:3,持续12h)中重结晶来将其纯化,得到v(0.84g,60%)。nmr谱与文献[2]中报道的数据一致。hplc(15min内50-100% b):tr=8.5min。
[0192][0193]
方案2sifa-ba的合成:a)tbdmsci、咪唑(dmf);b)tbuli、二-叔丁基二氟代硅烷(thf);c)hcl(meoh);d)氯铬酸吡啶盐(dcm);e)kmno4(dcm、叔丁醇、nah2po4缓冲液)。
[0194]
4)rhpsma-7.1-7.4的合成
[0195]
用于制备rhpsma-7的四种不同异构体的第一合成步骤为完全相同的,且采用上述标准fmoc-spps方案,从树脂结合的fmoc-d-orn(dde)-oh开始同时进行。在用20%哌啶于dmf中的溶液裂解fmoc基团(gp3)之后,使(tbuo)eue(otbu)2(2.0当量)与hoat(2.0当量)、tbtu(2.0当量)和dipea(6.0当量)于dmf中的溶液缀合4.5h。用2%肼/dmf的混合物裂解dde-基团(gp4a)之后,加入琥珀酸酐(7.0当量)和dipea(7.0当量)于dmf中的溶液并使之反应2.5h。通过向树脂中加入hoat(2.0当量)、tbtu(2.0当量)和dipea(6.0当量)于dmf中的混合物来实现fmoc-d-lys(otbu)
·
hcl(2.0当量)的缀合。预活化5min之后,加入溶于dmf的fmoc-d-lys(otbu)
·
hcl(2.0当量)并使之反应2.5h(gp2)。通过加入20%哌啶/dmf的混合物来进行后续fmoc-基团的裂解(gp3)。最后,将树脂分离以合成rhpsma-7.1-7.4(方案3)。
[0196]
rhpsma-7.1(d-dap-(r)-dota-ga):
[0197][0198]
在hoat(2.0当量)、tbtu(2.0当量)和2,4,6-三甲基吡啶(6.7当量)于dmf中的混合物中预活化fmoc-d-dap(dde)-oh(2.0当量),并加入的树脂结合肽中保持2.5h。使用溶于nmp和dmf的混合物中的咪唑和盐酸羟胺达3h来完成随后的正交dde-脱保护。使sifa-ba(1.5当量)与与作为活化试剂的hoat(1.5当量)、tbtu(1.5当量)和dipea(4.5当量)的侧链
的游离胺在dmf中反应2h。用哌啶进行fmoc-脱保护(gp3)之后,使(r)-dota-ga(tbu)4(2.0当量)与hoat(2.0当量)、tbtu(2.0当量)和2,4,6-三甲基吡啶(6.7当量)在dmf中缀合2.5h。依照gp5,在tfa中进行从树脂的裂解以及同时酸不稳定性保护基的脱保护。如在gp6中所述进行肽的
nat
ga-络合。
[0199]
rhpsma-7.2(l-dap-(r)-dota-ga):
[0200][0201]
在hoat(2.0当量)、tbtu(2.0当量)和2,4,6-三甲基吡啶(6.7当量)于dmf中的混合物中将fmoc-l-dap(dde)-oh(2.0当量)预活化2.5h。如对rhpsma-7.1所述进行随后的正交dde-脱保护、sifa-ba缀合和fmoc-裂解。使(r)-dota-ga(tbu)4(2.0当量)与hoat(2.0当量)、tbtu(2.0当量)和2,4,6-三甲基吡啶(6.7当量)于dmf中缀合2.5h。依照gp5,在tfa中进行从树脂的裂解以及同时酸不稳定性保护基的脱保护。如在gp6中所述进行肽的
nat
ga-络合。
[0202]
rhpsma-7.3(d-dap-(s)-dota-ga):
[0203][0204]
在hoat(2.0当量)、tbtu(2.0当量)和2,4,6-三甲基吡啶(6.7当量)于dmf中的混合物中将fmoc-d-dap(dde)-oh(2.0当量)预活化2.5h。如对rhpsma-7.1所述进行随后的正交dde-脱保护、sifa-ba缀合和fmoc-裂解。使(s)-dota-ga(tbu)4(2.0当量)与hoat(2.0当量)、tbtu(2.0当量)和2,4,6-三甲基吡啶(6.7当量)于dmf中缀合2.5h。依照gp5,在tfa中进行从树脂的裂解以及同时酸不稳定性保护基的脱保护。如在gp6中所述进行肽的
nat
ga-络合。
[0205]
rhpsma-7.4(l-dap-(s)-dota-ga):
[0206][0207]
在hoat(2.0当量)、tbtu(2.0当量)和2,4,6-三甲基吡啶(6.7当量)于dmf中的混合物中将fmoc-l-dap(dde)-oh(2.0当量)预活化2.5h。如对rhpsma-7.1所述进行随后的正交dde-脱保护、sifa-ba缀合和fmoc-裂解。使(s)-dota-ga(tbu)4(2.0当量)与hoat(2.0当量)、tbtu(2.0当量)和2,4,6-三甲基吡啶(6.7当量)于dmf中缀合2.5h。依照gp5,在tfa中进行从树脂的裂解以及同时酸不稳定性保护基的脱保护。如在gp6中所述进行肽的
nat
ga-络合。
[0208]
rhpsma-7.1:
[0209]
hplc(15min内10-70% b):tr=10.5min。
[0210]
hplc(40min内25-35% b):tr=31.4min。
[0211]
rhpsma-7.2:
[0212]
hplc(15min内10-70% b):tr=10.4min。
[0213]
hplc(40min内25-35% b):tr=27.9min。
[0214]
rhpsma-7.3:
[0215]
hplc(15min内10-70% b):tr=10.4min。
[0216]
hplc(40min内25-35% b):tr=28.1min。
[0217]
rhpsma-7.4:
[0218]
hplc(15min内10-70% b):tr=10.5min。
[0219]
hplc(40min内25-35% b):tr=29.1min。
[0220]
rhpsma-7.1-7.4:
[0221]
计算的单同位素质量(c63h96fgan12o25si):1536.6实测值:m/z=1539.4[m+h]+,770.3[m+2h]2
+
。
[0222][0223]
方案3rhpsma-7.1-7.4的合成:a)20%哌啶、(dmf);b)(tbuo)eue(otbu)2、hoat、tbtu、dipea、(dmf);c)2%肼(dmf);d)琥珀酸酐、dipea、(dmf);e)fmoc-d-lys(otbu)
·
hcl、hoat、tbtu、dipea、(dmf);f1)fmoc-d-dap(dde)-oh、hoat、tbtu、2,4,6-三甲基吡啶、(dmf);f2)fmoc-l-dap(dde)-oh、hoat、tbtu、2,4,6-三甲基吡啶、(dmf);g)咪唑、盐酸羟胺、(nmp、dmf);h)sifa-ba、hoat、tbtu、dipea(dmf);i1)(r)-dota-ga(tbu)4、hoat、tbtu、2,4,6-三甲基吡啶(dmf);i2)(s)-dota-ga(tbu)4、hoat、tbtu、2,4,6-三甲基吡啶(dmf);j)裂解和脱保护:tfa、tips、h2o;k)ga(no3)3、(tbuoh、h2o)。
[0224]
5)
18
f-标记
[0225]
对于
18
f-标记,采用了先前公布的程序,稍作修改。简单地说,使
18
f-水溶液通过用10ml水预处理的sax滤筒(sep-pak accell plus qma carbonate light)。用10ml空气干燥之后,通过用10ml无水乙腈接着用20ml空气冲洗滤筒来去除水。用溶于500μl无水乙腈中的100μmoloh-洗脱
18
f。标记之前,加入于无水乙腈(1m,30μl)中的30μmol草酸。该混合物作为整体或等分试样用于10-25nmol psma-sifa(1mm于无水dmso中)的氟化。将所得反应混合物于室温孵育5分钟。对于示踪剂的纯化,使用了先用10ml etoh接着用10ml h2o预处理的sep-pak c18 light滤筒。用9ml pbs(ph 3)稀释标记混合物并使之通过滤筒,接着通过10ml h2o。用500μl的etoh/水的4:1混合物(v/v)洗脱肽。通过放射性rp-hplc和放射性tlc(硅胶60rp-18f
254
s,流动相:补充有10%的2m naoac水溶液和1% tfa的mecn于h2o中的3:2混合物(v/v))来确定标记化合物的放射化学纯度。
[0226]
6)
125
i-标记
[0227]
根据先前公布的程序制备用于体外研究的参照配体([
125
i]i-ba)kue。简单地说,将0.1mg的甲锡烷基化前体(snbu
3-ba)(otbu)kue(otbu)2溶于含20μl过氧乙酸、5.0μl(21mbq)[
125
i]nai(74tbq/mmol,3.1gbq/ml,40mm naoh,hartmann analytic,braunschweig,德国)、20μl mecn和10μl醋酸的溶液中。将反应溶液于室温孵育10min,加载于滤筒上并用10ml水冲洗(c18 sep pak plus滤筒,用10ml meoh和10ml水预处理)。在用2.0ml的etoh/mecn的1:1混合物(v/v)洗脱之后,在缓氮气流下将放射性溶液蒸发至干燥并用200μl tfa处理30min,然后蒸发tfa。通过rp-hplc(20min内20%-40% b):tr=13.0min纯化([
125
i]i-ba)kue的粗产物。
[0228]
体外实验
[0229]
1)ic
50
的确定
[0230]
使psma-阳性lncap细胞在补充有10%胎牛血清并维持在37℃下加湿的5% co2气氛中的含glutamax-i的达尔伯克氏(dublecco)改良伊格尔(eagle)培养基/营养混合物f-12(1:1)(invitrigon)中生长。对于psma亲和力(ic
50
)的确定,在实验之前24
±
2小时收获细胞并将其接种到24-孔板中(1.5
×
105个细胞于1ml中/孔)。去除培养基之后,用500μl hbss(hank’s平衡盐溶液,biochrom,berlin,德国,添加了1%牛血清白蛋白(bsa))将细胞处理一次并置于冰上15min以在200μl hbss(1% bsa)中平衡。接着,加入每孔25μl溶液,所述溶液含hbss(1% bsa,对照)或浓度渐增的相应配体(在hbss中10-10-10-4
m),随后加入25μl于hbss(1% bsa)中的([
125
i]i-ba)kue(2.0nm)。对于每个浓度,所有实验均进行至少三次。在冰上孵育60min之后,通过去除培养基和用200μl hbss连续冲洗来终止实验。将两个步骤的培养基合并成一个流分并代表游离放射性配体的量。然后,用250μl的1m naoh溶解细胞并与后续洗涤步骤的200μl hbss合并。用γ-计数器完成结合放射性配体和游离放射性配体的定量。
[0231]
2)内化
[0232]
对于内化研究,在实验之前24
±
2小时收获lncap细胞并将其接种到24-孔板中(1ml/孔中1.25
×
105个细胞)。在去除培养基之后,用500μldmem-f12(5% bsa)将细胞洗涤一次并使之在200μl dmem-f12(5%bsa)中于37℃平衡至少15min。使用25μl的dmem-f12(5% bsa)或100μm pmpa溶液处理各孔进行封闭。接着,加入25μl的
68
ga/
18
f-标记的psma抑制剂(5.0nm)并使细胞在37℃孵育60min。通过将所述24-孔板置于冰上3min和连续去除培养基来终止试验。用250μl hbss冲洗各孔并合并来自前两步的流分,其代表游离放射性配体的量。通过用250μl冰冷的pmpa(10μm于pbs溶液中)溶液孵育细胞5min并用另外250μl冰冷的pbs再次冲洗来完成表面结合活性的去除。通过在250μl的1mnaoh中孵育细胞并与结合后续使用250μl 1.0m naoh洗涤的步骤的流分来确定内化活性。每次实验(对照和封闭)以一式三份进行。用γ-计数器定量游离的、表面结合的和内化的活性。所有内化研究均伴随使用([
125
i]i-ba)kue(c=0.2nm)以类似方式进行的对照研究。针对非特异性内化校正数据并针对放射性碘化的参比化合物所观察到的特异性内化进行归一化。
[0233]
3)辛醇-水分配系数
[0234]
在埃彭道夫(eppendorf)管中,将约1mbq的标记示踪剂溶于1ml的磷酸盐缓冲盐水(pbs,ph 7.4)和正辛醇的1:1混合物(按体积计)中。将混悬液于室温充分混合3分钟之后,以15000g将小管离心3分钟(biofuge15,heraus sepatech,osterode,德国)并用γ计数器
测定两个层的100μl等份。将所述试验重复至少六次。
[0235]
4)hsa结合
[0236]
对于hsa结合的确定,以0.5ml/min的恒定流速使用chiralpak hsa柱(50x 3mm,5μm,h13h-2433)。对每个试验新鲜制备流动相(a:nh4oac,50nm于水中,ph 7和b:异丙醇)且仅使用一天。使柱保持室温且每次运行均在检测到信号之后终止以减少采集时间。所有物质均以0.5mg/ml浓度溶于50%2-丙醇和50%50mm ph 6.9乙酸铵缓冲液中。鉴于假定了与肽有关的多种白蛋白结合,所选择的参照物质显示出从13%至99%的hsa结合范围。将全部九个参照物质连续注入以使用originpro 2016g建立非线性回归。
[0237]
表1:用于hsa-柱校准的参照物质。
[0238][0239]
保留时间是对所进行的试验的示例性显示;tr保留时间;lit.hsa人血清白蛋白结合的文献值,
[0240]
[%];log k has人血清白蛋白结合的对数k。
[0241]
体内试验
[0242]
所有动物试验均依照德国的一般动物福利法规和动物护理和使用机构指南进行。为建立肿瘤异种移植物,使lncap细胞(107个细胞/200μl)悬浮在含glutamax-i的dulbecco改良eagle培养基/营养混合物f-12(1:1)和基质胶(matrigel)(bd biosciences,德国)的1:1混合物(v/v)中并经皮下接种至6-8周龄cb17-scid小鼠(charles river,sulzfeld,德国)的右肩。当肿瘤生长至直径5-8mm(接种后3-4周)时使用小鼠。
[0243]
1)生物分布
[0244]
将约1-2mbq(《0.2nmol)的
18
f-标记的psma抑制剂注入到荷lncap肿瘤的雄性cb-17scid小鼠的尾静脉中并在注射之后1h将其处死(n=4-5)。取出选定器官,称重并用γ-计数器测定。
[0245]
2)代谢研究
[0246]
a)分析装置
[0247]
使用配备有spd-20auv/vis检测器(220nm、254nm)的shimadzu梯度系统(shimadzu deutschland gmbh,neufahrn,德国)进行分析型反相高压色谱(rp-hplc)。使用multospher 100rp18(125
×
4.6mm,5μm粒径)柱(cs gmbh,langerwehe,德国)以1ml/min的流速进行分析测定。用于所有hplc操作的洗脱液均为水(溶剂a)和乙腈(溶剂b),二者均含0.1%三氟乙酸。通过将uv-光度计的出口连接至herm lb 500检测器(berthold technologies gmbh,
bad wildbad,德国)来检测放射性。用于所有hplc操作的梯度均为:5% b等度0-3min,25-35% b 3-43min,95-95% b 43-48min。
[0248]
对于放射性薄层色谱,使用涂覆有硅胶60rp-18f
254
s的铝片,流动相由补充有10%的2m naoac水溶液和1% tfa的mecn于h2o中的3:2混合物(v/v)组成。使用scan-ram放射性tlc检测器(lablogic systems ltd.,sheffield,英国)或cr 35bio磷光成像仪(duerr medical gmbh,bietigheim-bissingen,德国)进行分析。
[0249]
b)rhpsma-7.1-7.4的代谢稳定性的确定
[0250]
对于体内代谢研究,将8-12mbq(《0.6nmol)的各
18
f-标记配体(rhpsma-7.1-7.4)注入到雌性健康cb17-scid小鼠的尾静脉中(n=4)。使小鼠保持麻醉30min并使用膀胱导管收集尿液。合并尿液样品并以9000rpm离心5min以去除悬浮物。直接使用上清液以上述条件进行放射性hplc分析。为证实在尿液中发生了
19
f与肽-结合
18
f的同位素交换,将各化合物与雌性健康cb-17-scid小鼠的尿液样品一起孵育特定的时间间隔,通过放射性hplc和/或放射性tlc对其进行分析。另外,该试验在添加过量的na
19
f(0.5μmol)并与
18
f-标记rhpsma-7.3一起孵育2h的情况下进行。
[0251]
c)rhpsma-7.1-7.4的体内分布的确定
[0252]
为定量各异构体(rhpsma-7.1-7.4)的相对摄取,将rhpsma-7的外消旋混合物(180-280mbq,sa=247-349gbq/μmol,在klinikum rechts der isar以全自动化程序产生)注射到荷瘤雄性cb-17-scid小鼠中。使动物保持麻醉30min并将其处死。收集并加工尿液、血液、肝脏、肾脏和肿瘤以进行下述程序。将尿液样品以9000rpm离心5min得到澄清溶液,并直接用于放射性hplc分析。用h2o将血液稀释至1ml并以13000g离心5min两次。收集上清液并加载至strata x滤筒(33μm聚合反相(polymeric reversed phase)500mg,用5ml meoh接着5ml h2o预处理)。用5ml h2o洗涤之后,用补充有1% tfa的mecn于h2o中的6:4混合物(v/v)洗脱滤筒。用水稀释洗脱物并通过放射性hplc进行分析。使用potter-elvehjem组织研磨器(kontes glass co,vineland,美国)或mm-400球磨机(retsch gmbh,haan,德国)将肿瘤、肾脏和肝脏均质化。
[0253]
i)potter-elvehjem组织研磨器
[0254]
将肿瘤和肾脏分别与1ml提取缓冲液(850μl 1m hepes ph7.4、100μl 20mm pmpa和100μl 1m nacl)一起在组织均质器中均质30min。收集所得均质物并于13000g离心5min。随后收集上清液,再次离心(13000g,5min)并加载至strata x滤筒(33μm polymeric reversed phase 500mg,用5ml meoh接着5ml h2o预处理)上。用5ml h2o洗涤之后,用补充有1% tfa的mecn于h2o中的6:4混合物(v/v)洗脱滤筒。用水稀释各器官的洗脱物并通过放射性hplc进行分析。
[0255]
ii)mm-400球磨机
[0256]
将器官(肿瘤、肾脏、肝脏)分别与3个研磨球(3mm直径)和1ml提取缓冲液(850μl 1m hepes ph7.4、100μl 20mm pmpa和100μl 1mnacl)一起在2ml管中于30hz均质10min。将均质物于13000g离心5min并收集上清液。随后,将沉淀悬浮于用1ml提取缓冲液中并用球磨机再次于30hz均质10min。离心分离(13000g,5min)之后,合并两次上清液并加载至strata x滤筒(33μm polymeric reversed phase 500mg,用5ml meoh接着5ml h2o预处理)上。用5ml h2o洗涤之后,用补充有1%tfa的mecn于h2o中的6:4混合物(v/v)洗脱滤筒。用水稀释各
器官的洗脱物并通过放射性hplc进行分析。为证实滤筒装载期间的穿漏并非未结合的f-18所致,亦通过放射性tlc检测离心分离之后的上清液。
[0257]
最后,从提取样品的hplc图谱确定各异构体的比率并与来自rhpsma-7的外消旋混合物的质量控制的异构体比率进行比较。经衰变校正的提取效率和滤筒装载效率以及试样的总提取活性在表2中给出。所有试验的滤筒洗脱效率均》99%。
[0258]
实施例2:结果
[0259]
色谱峰归属
[0260]
通过比较以下的uv图谱来进行色谱峰归属:
[0261]
a)rhpsma7-rac混合物与
[0262]
b)与各对应异构体纯的rhpsma7化合物共注入的rhpsma7-rac混合物。
[0263]
下列名称用于不同的异构体:
[0264]
rhpsma-rac:[
19
f][
nat
ga]d/l-dap-r/s-dotaga-rhpsma7rhpsma-7-1:[
19
f][
nat
ga]d-dap-r-dotaga-rhpsma7rhpsma-7-2:[
19
f][
nat
ga]l-dap-r-dotaga-rhpsma7rhpsma-7-3:[
19
f][
nat
ga]d-dap-s-dotaga-rhpsma7rhpsma-7-4:[
19
f][
nat
ga]l-dap-s-dotaga-rhpsma7
[0265]
表2:不同异构体、名称、典型保留时间(hplc条件在图2a-4b中给定)和每种异构体相在典型rhpsam7-rac混合物中的百分比的归属。每种异构体的确切的量可变化。
[0266][0267]
结合亲和力
[0268]
通过将在各配体的
nat
ga-络合之后直接获得的溶液用于稀释系列来确定第一组值(rhpsma-7.1和rhpsma-7.2;图6a)。在第二个数据组(图6b)中,通过rp-hplc纯化络合的配体以分离未络合的
nat
ga-盐。鉴于未观察到显著差异,合并了两个系列并用于计算平均值(
±
sd)。
[0269]
表3:个体ic
50
[nm]测量值(如在图6a和6b中所示)的描绘。条件如在对图6a的说明中所述。
[0270][0271]
*删除了rhpsma7.1系列的5号值(统计学离群值)。
[0272]
表4:其他选定psma抑制剂的结合亲和力(ic
50
[nm])(*)。
[0273][0274]
*在我们的实验室中使用完全相同的结合测定进行
[0275]
(robu等ejnmmi research 2018;8:30)。
[0276]
内化研究
[0277]
表5:个体内化率[[
125
i]ib-kue的%]的描绘。
[0278][0279]
表6:其他选定psma抑制剂的内化值[[
125
i]ib-kue的%](*)。
[0280][0281][0282]
*在我们的实验室中使用完全相同的结合测定进行
[0283]
(robu等ejnmmi research 2018;8:30)。
[0284]
亲脂性(辛醇-水分配系数)
[0285]
用磷酸盐缓冲盐水(pbs,ph 7.4)和正辛醇进行logp值的确定(=logp
oct/pbs
)。
[0286]
表7:在辛醇/pbs
7.4
混合物中确定的rhpsma7-异构体7.1-7.4异构体的个体log p
测量值。
[0287][0288]
表8:psma-1007、dcfpyl、rhpsma7-rac和rhpsam7.1-7.4异构体的log p值;(n=6),辛醇/pbs
7.4
。
[0289][0290]
psma抑制剂与人血浆蛋白的结合
[0291]
表9:psma-1007、dcfpyl、rhpsma7-rac和rhpsam7.1-7.4异构体的hsa结合;(n=
6)。在chiralpak hsa柱(50x 3mm,5μm,h13h-2433)上确定。
[0292][0293]
[
18
f][
nat
ga]rhpsma7.1-7.4在注射后1h的生物分布
[0294]
表10:
18
f-rhpsma在注射后1h在荷lncap肿瘤的scid小鼠中的生物分布(以%id/g计)。数据表示为平均值
±
sd(对于rhpsma7.1 n=4,对于rhpsma7.2 n=5,对于rhpsma7.3 n=4,对于rhpsma7.4 n=5和对于rhpsma7-rac n=3)。
[0295][0296][0297]
具有竞争的情况下[
18
f][
nat
ga]rhpsma7.1-7.4在腹腔注射(pi)后1h的生物分布
[0298]
表11:与pmpa(8mg/kg)共注射的
18
f-标记rhpsma示踪剂在腹腔注射后1h在lncap荷瘤scid小鼠中的生物分布[%id/g]。数据表示为平均值
±
sd(n=3)。
[0299][0300]
在施用[
18
f]rhpsma7-rac之后每种rhpsma7.x异构体在血液、肾脏、肝脏、尿液和肿瘤中的量的相对变化的定量
[0301]
出于在将[
18
f]rhpsma7-rac注入到荷lncap肿瘤小鼠之后30min对每种rhpsma7异构体在血液、肝脏、肾脏、尿液和肿瘤中的相对变化进行定量,使用两种不同的均质化方法(potter和球磨机)从肾脏、肝脏和肿瘤组织中提取示踪剂(参见材料和方法)。
[0302]
表12概述了对两种均质化方法观察到的效率和后续固相萃取程序(用以从蛋白流分中分离示踪剂)的功效。
[0303]
表12:经由potter-elvehjem组织研磨器(n=1)和mm-400球磨机(n=3)从实验组织试样确定衰变校正的提取活性。
[0304]
potter-elvehjem组织研磨器(n=1):
[0305][0306]
mm-400球磨机(n=3):
[0307][0308]
虽然使用potter从样品中提取活性相当有效,但球磨机的使用却令人失望。尽管如此,即便使用球磨机亦达到》60%提取效率。
[0309]
考虑可通过代谢裂解rhpsma7的酰胺键形成的可能的物质,似乎只有显著增加b)
f18-氟化物的亲脂性的a)物质才有可能。因此,原则上,在图12中所描绘的“il”物质似乎可能不是提取自组织样品(水溶液萃取)并因此未出现在最终分析中。然而,应注意的是,此类物质会在体内出现在肝脏和肠中(亲脂化合物的肝胆排泄)或者将与血浆蛋白结合(导致血液的高活性水平,这在另一方面显示优秀的提取效率)。
[0310]
为了对外消旋混合物中的每种异构体,尤其是对较差分离的第一峰和第二峰(rhpsma7.2和rhpsma7.3)进行定量,最先使用了图解近似法(graphical approximation)。该方法基于以下假设:a)各异构体以相同的峰形从hplc柱中洗脱和b)不同的峰高可用作一近似值以借由线性因子计算分离较差的峰(即rhpsma7.2和rhpsma7.3)。
[0311]
基于这些假设,通过使用与[
18
f][
nat
ga]rhpsma7-rac共注射的荷lncap肿瘤小鼠进行首个分析。出于借由另外三个试验来验证这些试验以及通过更有效的程序来改进所述图解分析的目的,使用了systat软件包
‘
peakfit’。peakfit允许通过去卷积程序进行hplc洗脱图谱的自动非线性分离、分析和定量,其使用具有傅里叶去卷积/滤波算法的高斯响应方程(https://systatsoftware.com/products/peakfit/)。
[0312]
第一个试验的图解分析的对比显示,图解分析过高估计了第二峰(rhpsma7.3),而过低估计了第一峰。因此,借由peakfit对所有数据组进行重新分析和定量。
[0313]
注射后30min在荷瘤小鼠中的4个独立试验的hplc-分析
[0314]
1.通过放射性hplc评价峰3和峰4(rhpsma7.4和rhpsma 7.1)
[0315]
首先检验去卷积技术是否对分离较好的(尽管未基线分离)后两个峰(rhpsma7.4和7.1)显示相似数据。
[0316]
2.通过放射性hplc评价所有峰(rhpsma7.1、rhpsma 7.2、rhpsma 7.3和rhpsma 7.4)
[0317]
图14a和14b概述了给定样品中每种rhpsam7.n异构体相对于其在注入溶液([
18
f][
nat
ga]rhpsma7-rac)中的百分比的百分数变化;独立试验的结果在图14中显示。通过systat
‘
peakfit’分析hplc洗脱图谱来定量各异构体的比例。然后,计算给定样品中每种异构体相对于其在注入溶液中的百分比的百分数变化。
[0318]
3.hplc数据讨论
[0319]
从均质化的(肾脏、肝脏、肿瘤)或稀释的(血液)组织中提取、然后固定在固相萃取滤筒上并自其洗脱的放射性的放射性hplc分析的确未显示代谢不稳定性的迹象。因此,未观察到亲脂的代谢片段。应注意的是,在用于样品制备的条件下,无法通过hplc来精确检测f-18-氟化物(参见tlc分析)。
[0320]
尽管有朝向d-dap-衍生物rhpsma7.1和rhpsma7.3的明显趋势,但总体变化较低(最高15%)。在本文中亦重点强调的是,图15和16是在不考虑绝对摄取值的情况下显示“相对变化”。
[0321]
尽管rhpsma7.1在所有rhpsma7化合物中具有最弱的亲和力和内化,但其在血液、肝脏、肾脏和肿瘤中显示最大的正向百分数变化。
[0322]
尽管该结果的原因还不清楚,但可推测的是,即便使用球磨机的组织样品的均质化,亦不会导致大量的细胞破碎。因此,具有最高内化的rhpsma7示踪剂(rhpsma7.2:191.83%
±
15.54%,rhpsma7.4:207.33
±
4.06%和rhpsma7.3:161.41%
±
8.88%)可能以低效的方式被提取,而仅具有69.55%
±
5.29%的低内化的rhpsma7.1被高效提取并因此在
hplc分析中被过高估计。
[0323]
此外,rhpsma化合物7.2和7.4似乎稍更迅速地排泄(参见尿的值)。尽管两种化合物与rhpsma7.1相比表现出更高的亲和力和内化率,但这些化合物通常在实体组织和血液中显示负变化。还不清楚这是否由7.2和7.4(二者均为l-dap衍生物)的代谢降解所致,因为未检测到代谢物,即亲液的代谢物。然而有可能的是,此类代谢物(参见图11)因其高logp值而在水性缓冲溶液中为不可提取的。在这种情况下,其应出现在肝脏样品中(参见生物分布)以及或许出现在血液样品中(对高血清蛋白结合的概率较高)。鉴于在生物分布研究期间对肝脏组织未观察到升高的活性蓄积以及从血液中提取活性为高效的(参见表3),我们假定的是,rhpsma7.2和rhpsma7.4未发生显著降解。在人体中临床使用[
18
f]rhpsma-rac的情况下肝脏组织(胆囊、肠)的不容怀疑的suv-值支持了这一假设。
[0324]
在注射后30min的荷瘤小鼠中的tlc-分析
[0325]
放射性tlc分析通过以下进行:a)通过直接将少量尿液样品置于tlc纸条(strip)上,b)通过分析少量的spe处理期间的非固定化活性物(
‘
穿透流分’),和c)通过分析少量的滤筒洗脱物。
[0326]
表13:血液、器官和尿液样品的tlc分析
[0327][0328][0329]
(*)因低活性水平所致,删除了信号强度《200cts的tlc测量值。
[0330]
tlc数据讨论
[0331]
鉴于非常难以借由rp-18色谱检测不添加载体的(n.c.a.)
18
f-氟化物(因基质上的游离si-oh基团与不添加载体的氟化物相互作用),因此进行薄层色谱研究以在提取的溶液
中定量f-18-氟化物。
[0332]
鉴于常用于蛋白质沉淀的试剂和盐均未进行冷氟化物测试,以及为了避免f-18-氟化物可能通过同位素交换从示踪剂中释放,因此在样品制备过程中未进行蛋白质沉淀——尽管这类蛋白质负载常常导致有限的峰分离、峰拖尾和活性滞留在起始线。将组织提取(或血液离心分离)之后获得的溶液直接用于tlc分析。
[0333]
尽管在所有样品中可进行分析的活性相当低,tlc结果显示f-18-氟化物在研究组织中的总含量均低于约6%,但以下除外:
[0334]-2018年7月30日获得的尿样(17.49%游离氟化物),
[0335]-2018年8月2日获得的肝脏样品(25.85%游离氟化物)。
[0336]
尽管通过tlc对尿液的分析被认为是有效结果(参见图20中的图谱),但使用肝脏样品获得的结果是因代表完整示踪剂的峰的严重(extensive)拖尾所致(参见图18)。此外,可推断上文提及的最高6%游离氟化物代表过高估计,因为对于临床应用,即便在qk期间和[f-18]rhpsma7-rac在pbs中释放期间获得的峰拖尾(图18)表明产物峰的拖尾。如通过磷光图像所证实,这种拖尾在几乎每个tcl分析中均观察到且贡献了f-18-氟化物的积分面积。
[0337]
需要注意的是,生物分布研究和人体临床pet扫描(2018年7月状态:约1400例使用[f-18]rhpsma7-rac扫描)均未因为释放的f-18-氟化物而导致在骨中的任何可疑的或可鉴定的f-18-蓄积。为进一步研究f-18氟化物从[f-18]rhpsma7-rac中的释放(如在一个尿样中观察到的),我们借由rp-18hplc(新型rp-18封端柱)和tlc分析研究了f-18-氟化物在另外的尿样(正常小鼠)中的出现。
[0338]
f-18-氟化物在注射后30min的正常小鼠中的形成的放射性tlc分析
[0339]
出于此目的,使用了正常小鼠。在30min时间段内借由导尿管收集尿液样品。将尿液离心并直接进行hplc和tlc分析。
[0340]
如在图21中左列所示,游离f-18-氟化物在所有异构体的尿液样品中均有发现且在新鲜尿液与[f-18]rhpsma7.4一起孵育时亦有形成(右列)。f-18-氟化物的鉴定通过证实以下来进行:a)该物质保留在qma滤筒上(未显示数据),b)在rp-18柱的死体积中洗脱和c)不论使用何用流动相,均不能分别在rp-18柱上或固定在rp-18tlc板上保留或移动。
[0341]
在血液或器官(例如肾脏、肿瘤、肝脏等)的hplc分析中未检测到如此大量的f-18氟化物,在小鼠的生物分布研究中未观察到在骨中活性摄取升高,以及在使用[f-18]rhpsma7-rac化合物的临床pet扫描期间未观察到在骨中活性摄取升高,因为已于2017年底在tum确定了[f-18]rhpsma7-rac用于临床扫描(2018年7月底状态:在患前列腺癌的患者中约1400例pet扫描),因为以上事实,我们推断[f-18]氟化物可能在示踪剂的肾小球过滤下游形成,导致[f-18]氟化物的形成和后续排泄,而在血液、器官或骨中的f-18-氟化物的摄取不可检测。
[0342]
描述肾脏和尿液中氟化物的相关量的关于氟化物毒理学的文献支持了该假设。在小鼠中观察到0.3ppm的正常尿氟化物水平(bouaziz h等,fluoride 2005;38(1):23-31)。在另一个出版物中,正常小鼠尿液中的平均氟化物浓度被确定为0.13-0.14μg/ml(poesina nd等rom j morphol embryol2014,55(2):343-349),以及inkielewicz i.等发现大鼠血清中的氟化物浓度为肾脏中氟化物浓度的约5%(血清:0.051μg/ml,肾脏:0.942μg/ml)(fluoride;36(4);263-266)。考虑到大部分示踪剂被肾脏特异性吸收并被肾生理清除,因
此在肾脏中氟化物水平升高,结合36.6℃的体温,可导致在肾脏中f-18-氟化物从rhpsma-化合物中的连续排出。
[0343]
因此,将从正常小鼠收集的新鲜的非放射性尿液样品与[f-18]rhpsma7.x一起孵育不同的时间(参见对图21的说明)。图22,右列,清楚证实尿液与[f-18]rhpsma7.x一起离体孵育会导致游离[f-18]氟化物不同程度的形成,该形成被尿液样品中不同浓度的冷f-19-氟化物促进的且随时间而增加。
[0344]
为进一步支持所述假设,向新鲜的非放射性小鼠尿液中加入500nmol冷f-19-氟化物,接着加入[f-18]rhpsma7.3并孵育2h。根据所述假设,高浓度的[f-19]氟化物将导致大量[f-18]氟化物的形成。图23显示在这些条件下,在2h内交换了98.5%的放射性并形成[f-18]氟化物(图23)。
[0345]
鉴于同位素交换率取决于平衡中的四种相关物质([f-18]氟化物、[f-19]氟化物、[f-18]rhpsma7.3和[f-19]rhpsma7.3)的浓度,亦研究了向新鲜的放射性尿液中加入[f-18]氟化物(20.6%[f-18]氟化物、79.4%[f-18]rhpsma7.3)随后加入冷[f-19]rhpsma7.3示踪剂是否亦会导致放射性药物[f-18]rhpsma7-3的标记。出人意料的是,在室温下,即便向上述尿液中加入5nm的少量[f-19]rhpsma7-3亦会导致[f-18]rhpsma7.3从79.4%增加到85.8%([f-18]氟化物从20.6%减少到14.2%)。
[0346]
认为通过尿中的同位素交换获得的结果代表了所有与4-(二-叔丁基[(18)f]氟代甲硅烷基)-苄基)氧基部分缀合的示踪剂,并因此代表了对于所有rhpsam7异构体。
[0347]
临床前剂量测定、人体生物分布和肿瘤病灶中的摄取
[0348]
请注意,在下文中,18f-rhpsma-7是指
nat
ga-18
f-rhpsma7-rac,18f-rhpsma-7.3是指
nat
ga-18
f-rhpsma7.3。
[0349]
a)小鼠中18f-rhpsma-7和18f-rhpsma-7.3的临床前剂量测定
[0350]
目的为评价在小鼠中单次静脉内施用之后
18
f-rhpsma-7和
18
f-rhpsma-7.3在直至300分钟的不同时间点的分布和排泄,以及对内部剂量测定进行计算。
[0351]
方法
[0352]
每个时间点分别用平均25.6
±
3.6mbq的18f-rhpsma-7和28.5
±
4.8mbq的18f-rhpsma-7.3注射3-5只小鼠。使用重度联合免疫缺陷(scid)小鼠进行试验。所有动物试验均依照德国的一般动物福利法规和针对动物护理和使用的机构指南进行。
[0353]
在下列时间点处死小鼠:
[0354]
18
f-rhpsma-7:施用之后10、20、40、60、120和180分钟。
[0355]
18
f-rhpsma-7.3:施用之后10、60、120、180-和300分钟。
[0356]
请注意,基于显示
18
f-rhpsma-7.3的延长肾脏摄取的初试验,将较迟的时间点(300min)用于终试验。
[0357]
收集下列组织/液体:
[0358]
尿液、血液、心脏、肺、脾、胰腺、肝脏、胃(排空的)、小肠(排空的)、大肠(排空的)、肾脏、膀胱、睾丸、脂肪、肌肉(部分股间肌)、股骨、尾部和脑。在co2气室中用移液器收集尿液。如果在气室中错过排尿,则用胰岛素注射器抽吸膀胱。在处死之后立即用胰岛素注射器从心脏抽血。所有其他组织和器官均进行解剖并直接转移到塑料容器中。
[0359]
使用电子天平测定塑料容器中样品的重量。预先测定用于专门样品的空的和预标
记的塑料容器的重量。从测量样品连同塑料容器的重量中减去所述塑料容器的皮重。如此计算的重量指定为测量样品的重量。
[0360]
将装有测量样品的塑料容器置于自动γ计数器(perkinelmer-wallac,waltham,美国)的特定样品架上以测定60秒内的计数率(每分钟计数=cpm)。此外,具有已知放射量的1%(v/v)标准品(n=5)与样品一起测量,以将器官样品的计数率转化成活性。
[0361]
数据分析
[0362]
针对衰变自动校正测量样品的计数率。使用下式确定测量样品中的放射性分布比(单位:注射剂量的百分数(%id))。将从一只小鼠获得的所有测量样品的计数率之和指定为所施用放射性的计数率。
[0363][0364]
每单位测量样品(不包括尿液和粪便样品)的重量的放射性分布数(单位:%id/g)通过使用下式来确定。通过从含有样品的容器中减去空的测量容器来确定测量样品的重量。
[0365][0366]
剂量测定分析
[0367]
为了每种放射性示踪剂的统计学计算的一致性,对于
18
f-rhpsma-7和
18
f-rhpsma-7.3,使用相同数量的时间点。因此,对于
18
f-rhpsma-7,将10min和20min时间点合并形成15min终点。
[0368]
根据j juan等,journal of pharmaceutical sciences,1993,82:762-763,使用数值积分和自然衰变二者来生成对重要源器官中蓄积的活性的时间积分(auc)。
[0369]
kirshner等建立了一种方法,其通过两个物种模型中器官重量和总体重的比率来线性缩放动物中的注射剂量百分数。
[0370]-kirschner as,ice rd,beierwaltes wh.131i-19-碘胆甾醇的辐射剂量学(radiation dosimetry of 131i-19-iodocholesterol).j nucl med.1973年9月1日;14(9):713-7。
[0371]-kirschner a,ice r,beierwaltes w.致编辑信.j nucl med.(1975):248-9。
[0372]
简单地说,为了从小鼠中的生物分布来计算人体剂量测定,需要外推法来解释动物和人之间的差异。使用在小鼠的生物分布研究中测得的时间依赖性器官活性浓度(以每克的注射剂量的百分数为单位,%id/g)和全身活性,来估计对70-kg标准成人解剖学模型的正常器官辐射剂量。
[0373]
使用标准成人和“标准”25-克小鼠中的相对分数器官质量,将小鼠中的组织活性浓度转换成70-kg标准成人中的组织分数活性。将时间依赖性全身活性拟合成指数函数,并假设注射活性和全身活性之间的差异会排泄到尿液中,因为肝脏和gi示踪剂中的活型浓度在研究的所有时间点均较低。
[0374]
采用梯形法则,通过数值积分计算器官停留时间,并将身体其余部位的
18
f停留时间计算为全身停留时间和器官与尿液停留时间之和之间的差异。在olinda/exm 1.0剂量学软件中使用动态排泄模型来评估膀胱内容物的停留时间。最后,使用olinda/exm 1.0计算
标准成人平均器官剂量当量(以msv/mbq计)和有效剂量(亦以msv/mbq计)。
[0375]
小鼠中的辐射吸收剂量和来自生物分布的剂量测定的最终计算:其中发生显著放射性蓄积的组织或器官(即,源器官)为肾脏、脾、肺、肝脏和心脏。对于活性蓄积和清除,发现从血液中清除并清除到尿中很快,但在肾脏中的积累相对较慢。
[0376]
结果
[0377]
表14.使用3.5h膀胱排泄间隔时
18
f-rhpsma-7的剂量测定结果。
[0378][0379]
表15.使用1.0h膀胱排泄间隔的
18
f-rhpsma-7的剂量学结果。
[0380][0381][0382]
表16.使用3.5h膀胱排泄间隔的
18
f-rhpsma-7.3的剂量学结果。
[0383][0384]
表17.使用1.0h膀胱排泄间隔的
18
f-rhpsma-7.3的剂量学结果。
[0385]
[0386][0387]
结论
[0388]
在小鼠中的所有检测时间点,在施用
18
f-rhpsma-7和
18
f-rhpsma-7.3二者之后的放射性分布比在肾中均为最高。此外,与其中活性比率低于8%id/g的所有其他评价的组织相比,两种放射性示踪剂在脾中和在膀胱中均较高。
[0389]
鉴于大部分
18
f-rhpsma-7/
18
f-rhpsma-7.3活性在肾脏中累加且经由膀胱的排泄物显示高放射性,故将主要排泄途径确定为经由肾脏和泌尿系统。
[0390]
使用3.5h和1.0h膀胱排泄间隔,外推的总有效剂量对于
18
f-rhpsma-7为2.66e-02msv/mbq和1.22e-02msv/mbq,且对于
18
f-rhpsma-7.3为2.17e-02msv/mbq和1.28e-02msv/mbq。假设1h排泄间隔的情况下,注射高达370mbq(10mci)用于临床扫描将得到两种试剂均小于5msv的令人满意的辐射照射。
[0391]
两种放射性示踪剂之间值得一提的差别仅在肾脏摄取方面明显,因为
18
f-rhpsma-7.3趋于随着保留时间的延长而逐渐地蓄积。然而,两种试剂的辐射照射为相当的。
[0392]
b)18f-rhpsma-7和18f-rhpsma-7.3的人体生物分布和在肿瘤病灶中的摄取
[0393]
以下章节描述了18f-rhpsma-7和18f-rhpsma-7.3的生物分布。在同情使用下进行概念验证评价。遵照德国药品法,amg
§
13 2b并遵从负责监管机构(上巴伐利亚行政区政府)应用药物。
[0394]
用biograph mct扫描仪(siemens medical solutions,erlangen,德国)检测所有受试者。以每个床位2-4min的采集时间用3d-模式获得所有pet扫描。针对随机性、无感时间、散射和衰减校正发射数据并通过有序子集期望最大化算法(四次迭代,八个子集)迭代重建,之后进行重建后平滑高斯滤波器(5-mm半峰全宽)。
[0395]
方法
[0396]
通过分别在47位患者和32位患者中分析临床
18
f-rhpsma-7-pet/ct和
18
f-rhpsma-7.3-pet/ct检验来评估人体生物分布。对于
18
f-rhpsma-7与
18
f-rhpsma-7.3,平均注射活性
分别为324(范围为236-424)mbq与345(范围为235-420)mbq,摄取时间分别为84(范围为42-166)min与76(范围为59-122)min。
[0397]
确定背景(臀肌)、正常器官(唾液腺、血池、肺、肝脏、脾、胰腺、十二指肠、肾脏、膀胱、骨)和三个代表性的肿瘤病灶的平均的和最大的标准化摄取值(suv平均值/suv最大值)。分别在89例病灶(26例原发肿瘤/局部复发、23例骨转移、38例淋巴结转移和2例内脏转移)和63例病灶(14例原发肿瘤/局部复发、30例骨转移、18例淋巴结转移和1例内脏转移)中分析了
18
f-rhpsma-7和
18
f-rhpsma-7.3的肿瘤摄取。
[0398]
对于suv的计算,在经轴切片中聚集增加的摄取的区域周围绘制圆形目标区域并自动适应50%等高线的三维目标体积(voi)。计算器官-背景比率和肿瘤-背景比率。
[0399]
结果
[0400]
18
f-rhpsma-7和
18
f-rhpsma-7.3的人体生物分布显示了从其他psma-配体已知的典型图谱。
18
f-rhpsma-7和
18
f-rhpsma-7.3的摄取参数非常相似,对于
18
f-rhpsma-7.3而言,在膀胱中的活性保留较低且在肿瘤病灶中的摄取较高:对于
18
f-rhpsma-7与
18
f-rhpsma-7.3来说,对于
18
f-rhpsma-7与
18
f-rhpsma-7.3的suv平均值分别为16.9与16.0(腮腺)、19.6与19.6(下颌下腺)、2.0与1.9(血池)、0.7与0.7(肺)、7.0与7.3(肝脏)、9.1与8.5(脾)、32.4与35.5(肾脏)、2.5与2.8(胰腺)、10.9与11.0(十二指肠)、1.1与1.3(未患病骨)和10.2与2.0(膀胱)。具体而言,
18
f-rhpsma-7与
18
f-rhpsma-7.3的摄取值对于在膀胱中的保留而言显著较低(2.0
±
0.8和6.3
±
21.2,p《0.05)并且对于肿瘤病灶而言显著较高(32.5
±
42.7vs.20.0
±
20.2,p《0.05)。
[0401]
表18.使用
18
f-rhpsma-7使正常器官和肿瘤病灶的suv最大值和suv平均值。数据显示为平均值、最小值和最大值。
[0402][0403]
表19.使用
18
f-rhpsma-7.3时正常器官和肿瘤病灶的suv最大值和suv平均值。数据显示为平均值、最小值和最大值。
[0404][0405][0406]
表20.使用
18
f-rhpsma-7时正常器官和肿瘤病灶的suv最大值和suv平均值与背景的比率。数据显示为平均值、最小值和最大值。
[0407][0408]
表21.使用
18
f-rhpsma-7.3时正常器官和肿瘤病灶的suv最大值和suv平均值与背景的比率。数据显示为平均值、最小值和最大值。
[0409][0410]
结论:
[0411]
对于大多数正常器官而言,
18
f-rhpsma-7和
18
f-rhpsma-7.3的人体分布相似。然而,对于
18
f-rhpsma-7.3而言,在膀胱中的示踪剂停留显著更低且在肿瘤病灶中的摄取显著更高,表现出用于临床成像的明显优势。图28中显示了具有
18
f-rhpsma-7.3的令人满意的人体生物分布和高肿瘤病灶摄取的成像实例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1