一种硼酸促进4-溴-7-甲基-1H-吲唑关环制备的合成方法与流程
本技术属于有机合成领域,具体涉及一种利用硼酸促进4-溴-7-甲基-1h-吲唑关环制备的合成方法。
背景技术:
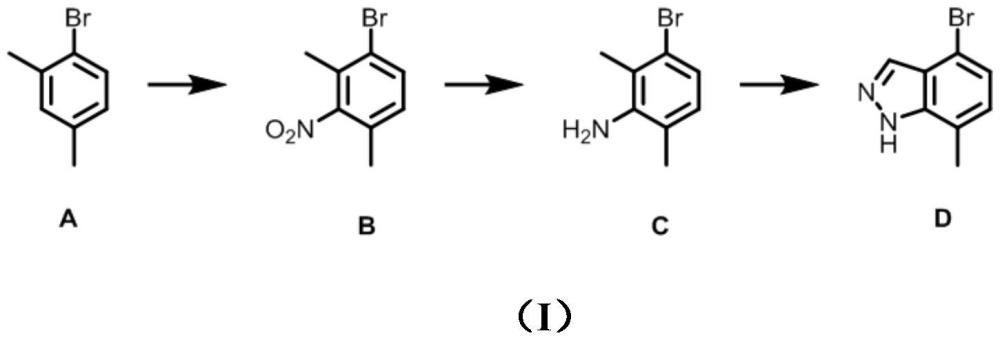
1、4-溴-7-甲基-1h-吲唑是一种重要的医药中间体,其在多种药物中作为重要分子砌块而广泛受到关注。现有技术中,4-溴-7-甲基-1h-吲唑的合成主要采用关环策略进行,例如主要通过重氮化反应关环反应制备,具体如下:
2、
3、上述反应中,以2,4-二甲基溴苯作为原料,进行硝化反应,继而将硝基还原得到化合物c,最后将化合物c的氨基转化为重氮盐后,在碱性条件下关环得到4-溴-7-甲基-1h-吲唑。该方法中,第一步需历经安全隐患大、设备要求高的硝化反应;并且,经实践,第一步与第三步的反应选择性较差,收率不理想;更为重要的是,目标产物d的合成过程中,产品与异构体性质非常接近,纯化困难。因此,现有技术中缺少一条路线简单、操作简便、安全隐患低、各步选择性高且收率高、后处理简单、适合工业化生产的4-溴-7-甲基-1h-吲唑合成路线。
技术实现思路
1、鉴于现有技术中4-溴-7-甲基-1h-吲唑合成方法收率低、后处理纯化难度大、实施安全隐患大、对设备要求高、难以放大工业生产等问题,发明人提供了一种合成4-溴-7-甲基-1h-吲唑的理想新方法。
2、本技术提供一种4-溴-7-甲基-1h-吲唑的制备方法m1,包含以下步骤:
3、
4、将6-溴-2-氟-3-甲基苯甲醛(以下称化合物2)溶于有机溶剂中,加入水合肼,升温下进行反应,加入硼酸后继续保温反应,经后处理得到目标化合物4-溴-7-甲基-1h-吲唑(以下称化合物3)。
5、在本发明的一个实施方案中,有机溶剂选自二甲基亚砜、二氧六环、氯仿、甲苯和乙醚中的一种或多种,优选为二甲基亚砜。
6、在本发明的一个实施方案中,有机溶剂与化合物2的体积质量比为2-10ml/g,优选为3-5ml/g。
7、在本发明的一个实施方案中,化合物2与水合肼、硼酸的摩尔比为为1:1.0-6.0:1.0-3.0,优选为1:5.5:1.5。
8、在本发明的一个实施方案中,反应温度为70-120℃,优选为90-110℃,更优选为100℃。
9、在本发明的一个实施方案中,升温下进行反应的反应时长为0.5-2h。
10、在本发明的一个实施方案中,加入硼酸后继续保温反应的反应时长为1-10h,优选为2-8h,更优选为3-5h。
11、在本发明的一个实施方案中,在加入硼酸之前,进行除去水和部分溶剂的操作,优选采用分水器对其进行分离。
12、在本发明的一个实施方案中,后处理步骤包括:将反应液加入水中进行淬灭,有机溶剂萃取后经减压蒸馏、纯化,制得化合物3。在本发明的一个实施方案中,用以萃取的有机溶剂选自乙酸乙酯、二氯甲烷、二氯乙烷和甲苯中的一种或多种,优选为乙酸乙酯。
13、在本发明的一个实施方案中,纯化步骤选自重结晶和柱层析中的一种或多种。在本发明的一个实施方案中,纯化步骤采用重结晶的方式进行,优选用乙酸乙酯与石油醚的混合液进行重结晶。更进一步地,乙酸乙酯与石油醚的体积比为1:1.2-2。
14、本技术还提供了一种4-溴-7-甲基-1h-吲唑的制备方法m2,包含以下步骤:
15、
16、步骤1、将2-氟-4-溴甲苯(以下称化合物1)溶于有机溶剂1中,在锂试剂存在下,与n,n-二甲基甲酰胺发生反应,经后处理得到化合物2;
17、步骤2、将化合物2溶于有机溶剂2中,加入水合肼,升温下进行反应,加入硼酸后继续保温反应,经后处理得到目标化合物3。
18、在本发明的一个实施方案中,步骤1在惰性气体保护下进行。在本发明的一个实施方案中,惰性气体为氩气或氮气。
19、在本发明的一个实施方案中,所述有机溶剂1选自乙腈、二甲基亚砜、二氧六环、四氢呋喃和乙醚中的一种或多种,优选为四氢呋喃或乙醚。
20、在本发明的一个实施方案中,所述锂试剂选自二异丙基氨基锂和正丁基锂中的一种或多种,优选为二异丙基氨基锂。
21、在本发明的一个实施方案中,有机溶剂1与化合物1的体积质量比为5-15ml/g。
22、在本发明的一个实施方案中,化合物1与锂试剂的摩尔比为1:1-1.5。
23、在本发明的一个实施方案中,化合物1与n,n-二甲基甲酰胺的质量体积比为1g/0.5-1ml。
24、在本发明的一个实施方案中,步骤1的反应温度为室温。
25、在本发明的一个实施方案中,步骤1的反应时长为0.5-3h。
26、在本发明的一个实施方案中,步骤1的后处理步骤包括:淬灭反应,有机溶剂萃取后进行减压蒸馏、纯化,制得化合物2。
27、在本发明的一个实施方案中,有机溶剂2选自二甲基亚砜、二氧六环、氯仿、甲苯和乙醚中的一种或多种,优选为二甲基亚砜。
28、在本发明的一个实施方案中,有机溶剂2与化合物2的体积质量比为2-10ml/g,优选为3-5ml/g。
29、在本发明的一个实施方案中,化合物2与水合肼、硼酸的摩尔比为1:1.0-6.0:1.0-3.0,优选为1:5.5:1.5。
30、在本发明的一个实施方案中,步骤2的反应温度为70-120℃,优选为90-110℃,更优选为100℃。
31、在本发明的一个实施方案中,步骤2中升温下进行反应的反应时长为0.5-2h。
32、在本发明的一个实施方案中,步骤2中加入硼酸后继续保温反应的反应时长为1-10h,优选为2-8h,更优选为3-5h。
33、在本发明的一个实施方案中,步骤2中,在加入硼酸之前,需进行除去水和部分溶剂的操作,优选采用分水器对其进行分离。
34、在本发明的一个实施方案中,步骤2的后处理步骤包括:将反应液加入水中进行淬灭,有机溶剂萃取后经减压蒸馏、纯化,制得化合物3。
35、在本发明的一个实施方案中,步骤1和步骤2中,用以萃取的有机溶剂选自乙酸乙酯、二氯甲烷、二氯乙烷和甲苯中的一种或多种,优选为乙酸乙酯;
36、在本发明的一个实施方案中,步骤1和步骤2中,所述纯化过程可以为柱层析纯化、重结晶纯化中的一种或多种。
37、在本发明的一个实施方案中,步骤1为:在惰性气体保护下,将化合物1加入有机溶剂1中,控温至-78℃,滴加锂试剂,保温搅拌1-3h,加入n,n-二甲基甲酰胺,升至室温,进行反应,反应完成后,淬灭反应,所得溶液用有机溶剂萃取,合并有机相,减压蒸馏除去溶剂,残余物经纯化得到化合物2。
38、在本发明的一个实施方案中,步骤2为:将化合物2加入有机溶剂2中,并于搅拌下,加入水合肼。加毕,升温反应0.5-2h,分离除去水和部分溶剂,加入硼酸,再继续保温反应1-5h,反应完全后,将反应液倒入水中,有机溶剂萃取,所得有机相减压蒸馏除去溶剂,所得残留物,经纯化得到目标化合物3。
39、本技术具有以下有益效果:
40、1、本发明提供了一条简便有效的合成路线,创造性地在关环步骤中,通过添加硼酸作为辅助试剂,促进关环反应正向进行,确保肟中间体能全部转化为产品,极大的提高了收率,避免了现有技术中异构体的生成,从而提高了反应收率,降低了产物纯化难度,为化合物的工业生成提供了可靠路线。
41、2、合成过程中,未采用硝化反应,避免了现有技术中,由于硝化而造成的高安全隐患,以及对反应设备的高要求;
42、3、本发明所用的原料和试剂均市售易得,后处理及纯化操作简便,适于放大化生产,为类似化合物的制备提供了可靠参考。
- 还没有人留言评论。精彩留言会获得点赞!