一种地屈孕酮中间体及其制备方法与流程
本发明涉及一种地屈孕酮中间体及其制备方法,属于药物和化学。
背景技术:
1、地屈孕酮(dydrogesterone,cas:152-62-5),化学名为9β,10α-孕甾-4,6-二烯-3,20-二酮,又称去氢孕酮,是由荷兰雅培公司生产的用于保胎及预防流产,以及由于内源性孕酮不足引起的各种疾病如痛经、子宫内膜异位症、继发性闭经、月经周期不规则、功能失调性子宫出血、经前期综合征、孕激素缺乏所致先兆性流产或习惯性流产、黄体不足所致不孕症等。目前有达芙通(duphaston,地屈孕酮片)和芬吗通(femoston,雌二醇片/雌二醇地屈孕酮片复合包装)两种产品在多个国家和地区销售。与天然孕激素(如黄体酮)相比,由于6,7位烯的存在和9,10位相反构型,在消化吸收和代谢过程中稳定,不易被破坏,可用于口服,且没有常见激素的副作用。地屈孕酮分子结构式如下:
2、
3、专利cn110198949b报道了一种全合成的方法制备地屈孕酮的方案,但是该方案中的起始原料需要多步合成,且会使用到剧毒试剂丙烯腈,整体路线效率比较低,不利于工业放大,反应式如下:
4、
5、专利wo2018109622报道了采用黄体酮为原料经过9步合成地屈孕酮,工艺中采用多步氧化还原反应及贵金属催化剂,总收率低于5%,不适合工业化生产。
6、
7、专利cn110818760b报道了以黄体酮为起始原料,经过羰基保护、溴化、消除、光照开环、光照合环、脱保护、异构化等步骤得到地屈孕酮。该路线的关键步骤在于两次光照反应,两步光照反应的收率只有30%左右,且所需设备复杂,不适于工业化生产。
8、
9、文献recueil des travaux chimiques des pays-bas(1971),90:27-32报道了以黄体酮为原料,通过乙二醇对双酮的保护、溴化、脱溴、高压汞灯光照再重排得到地屈孕酮的路线。这条路线中乙二醇的保护收率比较低(32-67%the journal oforganicchemistry,1952,vol.17,p.1369,1373),溴化和脱溴也存在很多的异构导致两步收率比较低(49%),在关键的光照步骤收率也仅仅为22%,不利于工业化生产。
10、
11、专利cn114957369b报道了如下制备地屈孕酮的路线,该路线经过光化学、生物发酵、双键转移、氧化、烯胺化、氧化6步得到最终产物。路线中光化学和生物发酵两步收率仅达7%,导致整条路线收率较低,不利于工业放大,反应式如下:
12、
13、专利cn114957372b报道了如下制备地屈孕酮的路线,该路线经过光化学、氧化、双键转移、水解、氧化、烯胺化、氧化7步得到最终产物。路线中光化学收率在加滤光剂的情况下仅达24.5%,不加滤光剂收率仅9%,收率较低,不利于工业放大,反应式如下:
14、
15、目前地屈孕酮的合成主要有两个思路,1)采用9,10位构型与地屈孕酮相同的孕甾烷类化合物为底物,构建20位羰基,存在的问题是步骤长,操作复杂,导致最终产率低;2)通过光反应构建9,10位构型,其中难点是双键移位构建5,7-二烯和光反应过程,存在的问题是5,7-二烯不易实现或者原子经济性差,光反应收率不高等。
16、因此,目前急需要开发一种新的地屈孕酮的制备方法。
技术实现思路
1、为了解决上述报道中存在的技术问题,本发明提供了一种合成地屈孕酮的新方法,以及新的地屈孕酮中间体及其制备方法。
2、为克服现有技术缺陷,实现商业化放大生产要求,本发明采用以下优选技术方案:
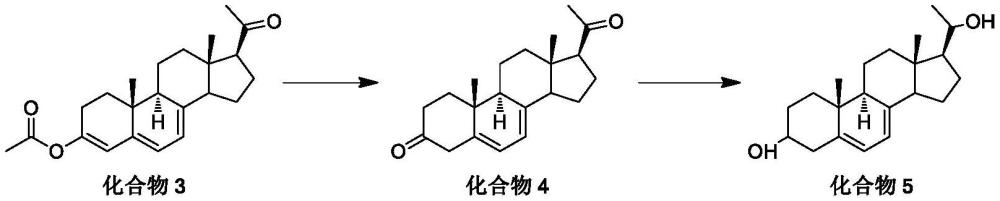
3、本发明第一方面提供一种式5化合物的制备方法,反应式如下所示:
4、
5、包括如下步骤:
6、(1)将式3化合物,在有机溶剂和无机碱存在下,进行水解反应得到式4化合物;
7、(2)将式4化合物进行还原反应得到式5化合物;
8、作为本发明的进一步改进,所述步骤(1)中无机碱选自碳酸钾、碳酸氢钾、碳酸钠、碳酸氢钠、氢氧化钾、氢氧化锂、氢氧化钠、碳酸铯等中的一种或多种;
9、作为本发明的进一步改进,所述步骤(1)式3化合物与无机碱的摩尔比为1:(1~5),优选为1:(1.5~3);
10、作为本发明的进一步改进,所述步骤(1)的有机溶剂选自甲醇、乙醇、异丙醇、正丁醇、四氢呋喃、1,4-二氧六环、乙腈中的一种或多种;
11、作为本发明的进一步改进,所述步骤(1)的有机溶剂的体积用量(ml)为式3化合物质量用量(g)的2~10倍,优选为4~6倍;
12、作为本发明的进一步改进,所述步骤(1)水解反应的温度为15~45℃;
13、作为本发明的进一步改进,所述步骤(1)水解反应的反应时间为1~5h;
14、作为本发明的进一步改进,所述步骤(1)水解反应反应完全后,过滤滤除无机碱(例如碳酸钾),滤液减压浓缩,剩余物溶于水,稀盐酸调ph至5-6,用乙酸乙酯萃取、无水硫酸钠干燥,过滤,浓缩,得到化合物4。
15、作为本发明的进一步改进,所述步骤(2)还原反应包含式4化合物,在醚类溶剂或腈类溶剂与醇类溶剂的混合溶剂中,还原剂存在下反应;
16、作为本发明的进一步改进,所述步骤(2)的还原剂选自硼氢化钠、硼氢化钾、硼氢化锂、硼氢化锌、硼氢化铝、硼烷等中的一种或多种;
17、作为本发明的进一步改进,所述步骤(2)式4化合物与还原剂的摩尔比为1:(1~5),优选为1:(1.1~2);
18、作为本发明的进一步改进,所述步骤(2)的醚类选自四氢呋喃、乙醚一种或组合;
19、作为本发明的进一步改进,所述步骤(2)的腈类溶剂选自乙腈;
20、作为本发明的进一步改进,所述步骤(2)的醇类溶剂选自甲醇、乙醇、异丙醇一种或组合;
21、作为本发明的进一步改进,所述步骤(2)的醚类溶剂或腈类溶剂的体积用量(ml)为式4化合物质量用量(g)的1~10倍,优选为3~5倍;
22、作为本发明的进一步改进,所述步骤(2)的醚类溶剂或腈类溶剂的体积用量(ml)为醇类溶剂的体积用量(ml)的1~10倍,优选为1~3倍;
23、作为本发明的进一步改进,所述步骤(2)还原反应的反应温度为-20~10℃,优选为-15~0℃;
24、作为本发明的进一步改进,所述步骤(2)还原反应的反应时间为5~18h,优选为10~15h;
25、作为本发明的进一步改进,所述步骤(2)反应完全后,将反应液缓慢倒入冰水中,随后缓慢滴加冰醋酸,析出的固体后,抽滤,用水淋洗滤饼;可选择性地再将滤饼溶于二氯甲烷中,用水和饱和食盐水洗涤,有机相减压浓缩,剩余物溶于甲醇,减压蒸馏,待剩余混合物体积至1/6~1/3,放于0~5℃析晶,抽滤,选择性地使用冷甲醇洗涤,滤饼55-60℃鼓风烘干,得到化合物5。
26、本发明第二方面提供一种式6化合物的制备方法,反应式如下所示:
27、
28、包括如下步骤:
29、(1)将式3化合物,在有机溶剂和无机碱存在下,进行水解反应得到式4化合物;
30、(2)将式4化合物进行还原反应得到式5化合物;
31、(3)将式5化合物进行光化学反应得到式6化合物。
32、包括以下方法:
33、步骤(1)和(2)的制备方法引用上述第一方面所有技术方案。
34、作为本发明的进一步改进,所述步骤(3)光化学反应,包含式5化合物,在有机溶剂中,led灯和高压汞灯的光源的照射下,碱存在下进行光化学反应,使c-10位的甲基由β构型翻转为α构型;
35、作为本发明的进一步改进,所述步骤(3)所述光化学反应分两个阶段进行,第一阶段在200-300nm波长范围的led灯照射下发生开环,第二阶段在300-400nm波长范围的高压汞灯下发生闭环;优选第一阶段led灯照射下发生开环的主波长为254nm,第二阶段高压汞灯灯下发生闭环的主波长为365nm;
36、作为本发明的进一步改进,所述步骤(3)光化学反应的第一阶段或第二阶段的反应时间为5~15h,优选8~12h;
37、作为本发明的进一步改进,所述步骤(3)的有机溶剂选自甲苯、甲醇、乙醇、四氢呋喃、甲基叔丁基醚、乙酸乙酯、甲酸乙酯、二氧六环、乙腈中的一种或几种;
38、作为本发明的进一步改进,所述步骤(3)光化学反应的温度为0~40℃,优选5~25℃;
39、作为本发明的进一步改进,所述步骤(3)的碱选自三乙胺,二异丙基乙基胺,三异丙胺、吡啶、三甲基吡啶等中的一种或多种;
40、作为本发明的进一步改进,所述步骤(3)的碱的体积用量(ml)为有机溶剂的体积用量(l)的2~10倍,优选为4~6倍;
41、作为本发明的进一步改进,所述步骤(3)中有机溶剂的体积用量(l)为式6化合物质量用量(g)的0.1~0.5倍,优选为0.2~0.3倍;
42、作为本发明的进一步改进,所述步骤(3)光化学反应反应完成后,将反应液减压蒸干溶剂,然后加入乙醇,配制成质量浓度为2.3%至3%的悬浮液,过滤,将得到的澄清溶液置入5℃以下析晶6小时,过滤,得到固体化合物6。
43、本发明第三方面提供一种式3化合物的制备方法,反应式如下所示:
44、
45、将式2化合物,在乙酰氯和乙酸酐存在下,进行双键转移反应得到式3化合物;
46、作为本发明的进一步改进,所述双键转移反应式2化合物与乙酰氯的摩尔质量比为1:(1~10),优选1:(1.1~5),例如1:4.4;
47、作为本发明的进一步改进,所述双键转移反应乙酸酐的体积用量(ml)为式2化合物质量用量(g)的3~10倍,优选为4~8倍;
48、作为本发明的进一步改进,所述双键转移反应的反应温度为30~80℃,优选40~60℃;
49、作为本发明的进一步改进,所述双键转移反应的反应时间为5~20小时,优选为6~15小时,再优选8~12小时;
50、作为本发明的进一步改进,所述双键转移反应反应完成后,反应液冷至室温,反应液在75℃左右减压浓缩,放置室温后滴加甲醇,再加入丙酮,减压旋去溶剂,剩余物再次溶于丙酮,减压浓缩至混合物体积至1/3~1/2,放于0~5℃析晶,2小时后过滤,洗涤,滤饼55-60℃烘干,得化合物3。
51、本发明第四方面提供一种新关键中间体式3化合物,结构如下:
52、
53、本发明第五方面提供所述式3化合物在制备地屈孕酮或者其中间体中的应用,具体一种地屈孕酮的制备方法,包含步骤如下:
54、
55、包括如下步骤:
56、(1)将式2化合物,在乙酰氯和乙酸酐存在下,进行双键转移反应得到式3化合物;
57、(2)将式3化合物,在有机溶剂和无机碱存在下,进行水解反应得到式4化合物;
58、(3)将式4化合物进行还原反应得到式5化合物;
59、(4)将式5化合物进行光化学反应得到式6化合物;
60、(5)将式6化合物在有机溶剂中,四甲基哌啶氧化物(tempo)、溴化钾和次氯酸钠体系条件下进行氧化反应得到式7化合物;
61、(6)将式7化合物在有机溶剂中,叔丁基对苯二酚、盐酸乙醇中进行双键转移反应得到地屈孕酮。
62、包括以下方法:
63、步骤(1)~(4)的制备方法引用上述第一、二、三方面所有技术方案。
64、作为本发明的进一步改进,所述步骤(5)氧化反应,包含如下步骤:将化合物7溶于二氯甲烷中,加入四甲基哌啶氧化物(tempo)和溴化钾,保持0~5℃搅拌,于该温度下滴加7.5%次氯酸钠水溶液,加完后搅拌30分钟。
65、作为本发明的进一步改进,所述步骤(5)氧化反应反应完成后,加入硫代硫酸钠饱和溶液淬灭,有机相用饱和硫代硫酸钠和3%稀盐酸水各洗涤一次,有机相无水硫酸钠干燥,过滤,浓缩,得到的粗品溶于正庚烷和甲基叔丁基醚的混合溶剂中(体积比3:1),0~5℃析晶1小时,抽滤,滤饼用正庚烷洗涤,滤饼在55~60℃鼓风烘干,得到式7化合物。
66、作为本发明的进一步改进,所述步骤(6)双键转移反应,包含如下步骤:将化合物8溶于二氯甲烷中,加入叔丁基对苯二酚,搅拌,氮气保护下,于0~10℃将质量分数35%盐酸乙醇溶液滴入,滴完后,保持0~10℃下反应2小时。
67、作为本发明的进一步改进,所述步骤(6)双键转移反应反应完成后,加去离子水淬灭反应,有机相用碳酸氢钠饱和溶液洗涤至中性,有机相浓缩,再用乙醇溶解,浓缩,反复1~3次,再浓缩至约混合物体积至1/3~1/2,放于-20℃析晶并冷冻2小时,过滤,滤饼用冷乙醇洗涤,滤饼再悬浮于正庚烷打浆2小时,过滤,用正庚烷洗涤,滤饼在55-60℃鼓风烘干,得到地屈孕酮。
68、与现有技术相比,本发明具有以下有益效果:
69、本技术以廉价易得的黄体酮为起始原料,经氧化、双键移位、酯水解、还原、构型转换、氧化、双键移位得到地屈孕酮;具有合成路线简短、成本低、纯化简单,收率高、避免有毒试剂的使用等优点;易于实现工业化大生产。
70、本技术化合物4和化合物5制备的两步摩尔收率70%,现有技术两个羰基同时还原收率仅60%,不同底物碳酸钾水解收率最高也仅85%;本技术相对于现有技术有明显技术优势。
71、本技术重复湖南科瑞生物制药cn112608361b实施例无水氯化钙、吡啶+硼氢化钠条件,收率也仅53.8%,因为该条件并不适用本技术底物,难以获得技术优势。
72、化合物7光反应,现有技术不另外添加抗氧化剂、促进光反应剂、微反应器、滤光等,光反应均不足收率30%,本技术收率达到39%,收率提高9%。
- 还没有人留言评论。精彩留言会获得点赞!