一种沙库巴曲关键中间体的制备方法与流程
本发明属于医药中间体合成,具体涉及一种沙库巴曲关键中间体(2r,4s)-5-(联苯-4-基)-4-[(叔丁氧羰基)氨基]-2-甲基戊酸的制备方法。
背景技术:
1、脑啡肽酶抑制剂沙库巴曲(sacubitril),化学名为4-(((2s,4r)-1-([1,1'-联苯]-4-基)-5-乙氧基-4-甲基-5-氧代丙烷-2-基)氨基)-4-氧代丁酸,其结构如下:
2、
3、复方制剂沙库巴曲缬沙坦钠片(entresto)是由诺华公司开发的双效血管紧张素受体-脑啡肽酶抑制剂,可用于高血压和心力衰竭的治疗。其有效降低慢性心力衰竭(nyhaⅱ~ⅳ级)患者因心衰而住院和死亡的风险。
4、沙库巴曲关键中间体氨基为boc保护基时,(2r,4s)-5-(联苯-4-基)-4-[(叔丁氧基羰基)氨基]-2-甲基戊酸(cas:1012341-50-2),结构式如下:
5、
6、目前化合物ⅰ的工艺主要是按照wo2008031567a1、wo2014032627a1合成路线制备。以4-溴联苯作为联苯引入试剂,通过格氏反应,与s-环氧氯丙烷形成联苯类化合物,再通过光延反应(mitsunobu反应)形成手性氨基类醇。将此醇氧化形成醛基后,维蒂希反应(witting反应)形成烯键,并水解酯基。运用金属加氢催化得到产品(2r,4s)-5-([1,1联苯基]-4-基)-4-((叔丁氧羰基)氨基)-2-甲基戊酸,本方法路线步骤长,总收率低,合成成本较高。其反应式如下所示:
7、
8、专利wo2008083967a3公开以l-焦谷氨酸为起始原料,通过缩合形成酰胺键后,用联苯类格式试剂对底物构筑联苯基团。之后通过还原及氨基保护后,在五环羰基α位手性选择形成甲基,再经过脱保护、开环、boc氨基保护后形成产品(2r,4s)-5-([l,1-联苯基]-4-基)-4-((叔丁氧羰基)氨基)-2-甲基戊酸,该方法反应条件需要低温,反应收率低,成本较高。其反应式如下所示:
9、
10、综上所述,现有报道的制备工艺中,沙库巴曲关键手性中间体i的制备,因反应条件苛刻、合成路线长、收率低、生产成本高、操作繁琐而不利于工业化生产。因此,有必要开发出关键手性中间体i更简便、经济和便于工业化的生产路线,工业上具有重要意义。
技术实现思路
1、本技术为解决现有技术存在制备沙库巴曲中间体路线冗长、收率低、手性异构体纯度低、和成本高等问题,开发了一种新的沙库巴曲中间体合成路线。该路线操作简单,反应收率较高,避免使用成本较高的催化剂和配体,产物手性纯度高,适合于工业化推广应用。
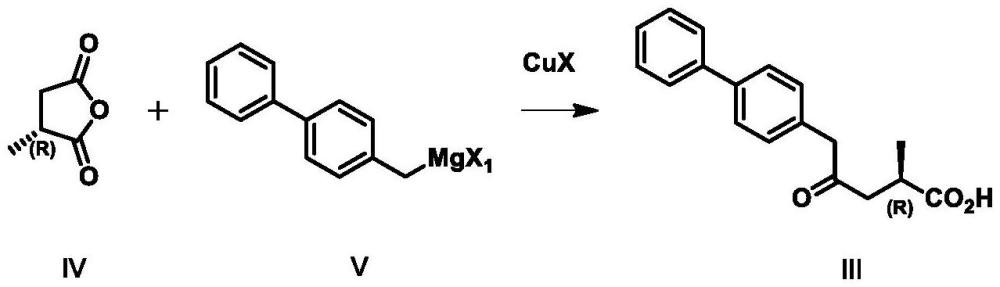
2、本技术第一方面提供式ⅲ化合物的制备方法,包括将式ⅳ化合物与式v化合物,在cux作用下进行开环反应,其反应式如下所示:
3、
4、其中,x和x1分别独立选自cl、br、i;
5、作为本技术的进一步的改进,所述式ⅳ化合物与式v化合物的摩尔比为1:(1~5),例如1:(1~3);
6、作为本技术的进一步的改进,所述ⅳ化合物与cux的摩尔比为1:(0.03~0.3),例如1:(0.03~0.2),再例如1:0.045;
7、作为本技术的进一步的改进,所述式v化合物1,1’-联苯-4-甲基卤化镁可选自1,1’-联苯-4-甲基氯化镁、1,1’-联苯-4-甲基溴化镁或1,1’-联苯-4-甲基碘化镁中的任意一种,例如1,1’-联苯-4-甲基溴化镁;
8、作为本技术的进一步的改进,所述cux选自cui、cubr、cucl中的任意一种,例如cui;
9、作为本技术的进一步的改进,所述开环反应是在有机溶剂中进行,所述有机溶剂选自醚类、芳香烃类中的至少一种,所述醚类为环戊基甲醚、四氢呋喃、2-甲基四氢呋喃、甲基叔丁基醚、1,4-二氧六环、丙醚、异丙醚、乙醚、乙二醇二甲醚或苯甲醚中的任意一种,所述芳香烃类为甲苯或二甲苯中任意一种;
10、作为本技术的进一步改进,所述有机溶剂的体积用量(ml)是式ⅳ化合物质量用量(g)的1~10倍,例如5~8倍;在本技术的一些具体实施方式中,所述有机溶剂的体积用量(ml)是式ⅳ化合物质量用量(g)的5倍;
11、作为本技术的进一步改进,所述开环反应温度为-30~0℃,例如-25~-15℃;所述反应时间为1~24h,例如1~12h;在本技术的一些具体实施方式中,所述反应温度为-25~-15℃,反应时间为1h;
12、在本技术的一些具体实施方式中,提供式ⅲ化合物的制备方法,包括:
13、1)在惰性氛围下,加入溴乙烷、镁屑,45~55℃滴加约1/20部分量4-溴甲基联苯的四氢呋喃溶液,确认引发后,继续保持45~55℃滴加剩余量约总量19/20的4-溴甲基联苯的四氢呋喃溶液;
14、2)滴加完成后继续搅拌反应1小时,降温至-15~-25℃,加入碘化亚铜,继续滴加化合物ⅳ的四氢呋喃溶液,滴加完后进行-15~-25℃保温搅拌1小时制备得到式ⅲ化合物。
15、作为本技术的进一步改进,所述开环反应通过简单分离得到式ⅲ化合物;
16、本技术未对分离步骤特别限定,可以采用本领域已知的分离步骤,只要能实现本发明的目的即可。例如,分离步骤可以包括但不限于:淬灭、萃取、洗涤、干燥、浓缩、重结晶和过滤等。在分离步骤中用到的溶剂可以采用本领域已知的常规溶剂。
17、在本技术的一些具体实施方式中,可选择性将式ⅲ化合物进行后处理,包括开环反应反应完成后,将反应液缓慢倒入稀盐酸中,10~30℃搅拌0.5h。然后在反应瓶中加入乙酸乙酯萃取,水相再用乙酸乙酯萃取,合并有机相,将有机相减压浓缩至1/5~1/2体积,降温至0~10℃抽滤、干燥后得到化合物iii固体。
18、本技术的联苯化合物由现有技术常规的制备方法得到,例如所述的1,1’-联苯-4-甲基溴化镁参考期刊文献(journal of the american chemical society(2017),139(37),13126-13140)的方法制备得到或本发明实施例1得到。
19、本技术第二方面提供式i化合物的制备方法,包括将本技术第一方面制备得到式ⅲ化合物作为原料,经过不对称还原胺化反应和上氨基保护基反应得到式i化合物;
20、
21、r为氨基保护基,例如叔丁氧羰基,苄氧羰基、芴甲氧羰基、对甲氧基苄基、苄基、烯丙氧羰基中的任意一种;
22、作为本技术的进一步改进,在手性钌催化剂作用下,化合物iii与氨源和氢气条件下,经过不对称还原胺化反应下得到式ii化合物,
23、作为本技术的进一步改进,所述氨源选自氨气、氨水、氨气溶液、醋酸铵、甲酸铵、氯化铵、硫酸铵、碳酸铵中的一种或多种;
24、作为本技术的进一步改进,所述氨源选自氨气溶液时,可以是氨气的甲醇、乙醇、异丙醇、乙腈、四氢呋喃或1,4-二氧六环、乙酸乙酯溶液中的任意一种(例如氨甲醇溶液,氨甲醇溶液摩尔浓度为4mol/l~7mol/l);
25、所述化合物iii与氨源的摩尔比为1:1~20;
26、作为本技术的进一步改进,所述手性钌催化剂例如ru(oac)2[(s)-meo-biphep]或(s)-ru(oac)2(binap);
27、作为本技术的进一步改进,所述式ⅲ化合物与钌催化剂的摩尔比选自1:0.0001~0.001;例如1:0.0003~0.001,再例如1:0.00035;
28、作为本技术的进一步改进,所述氢气压力范围选自0.5~10mpa,优选1.5~2.5mpa;
29、作为本技术的进一步改进,所述不对称还原胺化的反应温度选自45~70℃,例如55~65℃;
30、作为本技术的进一步改进,所述不对称还原胺化的反应时间选自12~48h,例如48h;
31、作为本技术的进一步改进,所述不对称还原胺化的反应溶剂选自甲醇、乙醇、异丙醇、乙腈、四氢呋喃、1,4-二氧六环、乙酸乙酯中的任意一种,例如甲醇;
32、作为本技术的进一步改进,所述不对称还原胺化反应完成后,可选择性地将反应液冷却至室温,氮气置换后,滴加氯化氢甲醇溶液至ph=2~3,搅拌1h,降温至0~10℃过滤,洗涤,干燥,得到化合物固体盐酸盐。
33、作为本技术的进一步改进,上氨基保护基反应可通过保护基需要选择合理的反应条件进行;例如化合物ii或其盐,在碱作用下,与氨基保护基试剂反应。
34、作为本技术的进一步改进,所述上氨基保护基反应中,化合物ii或盐与氨基保护基试剂的摩尔比为1:1~1.2;反应温度为50~60℃;反应时间为2~12h;
35、作为本技术的进一步改进,所述上氨基保护基反应的溶剂选自水、甲醇、乙醇、异丙醇、二氯甲烷、乙腈、四氢呋喃、1,4-二氧六环、乙酸乙酯中的一种或其任意组合,例如甲醇和水。
36、作为本技术的进一步改进,所述碱选自有机碱或无机碱,有机碱选自三乙胺、二乙胺、吡啶、二异丙基乙基胺中的任意一种,例如二异丙基乙基胺;无机碱选自磷酸钾、醋酸钾、碳酸钾、氢氧化钾、磷酸钠、醋酸钠、碳酸钠、氢氧化钠、碳酸氢钾、碳酸氢钠、氢氧化锂、和水合氢氧化锂中的一种或多种,例如氢氧化钠;
37、作为本技术的进一步改进,所述反应中化合物ii与碱的摩尔比为1:1.1~5,例如1:2;
38、在本技术的一些具体实施方式中,例如r为boc保护基,当r为boc氨基保护基,氨基保护基试剂选自二碳酸二叔丁酯,本发明提供式ia化合物的制备方法,包含:
39、
40、将化合物ii盐酸盐,加入甲醇和水,然后加入碱(例如氢氧化钠),控制50~60℃滴加二碳酸二叔丁酯,滴完继续保温(例如2小时),蒸馏浓缩,过滤,减压干燥,得到式ia化合物。
41、本技术第三方面提供沙库巴曲(sacubitril)的制备方法,包括本发明第一方面所述制备方法得到式ⅲ化合物,再由式ⅲ化合物制备得到沙库巴曲。
42、本技术第四方面提供沙库巴曲(sacubitril)的制备方法,包括本发明第二方面所述制备方法得到式i化合物,再由式i化合物制备得到沙库巴曲。
43、与现有技术相比,本发明的有益技术效果在于:
44、(1)本发明通过手性原料式ⅳ化合物直接引入手性2-甲基戊酸基团,反应路线短,节省时间,同时提高了手性异构体纯度,后处理操作简便。
45、(2)本发明优化了工艺,操作简单,原料可及性高,显著降低成本,适合工业化生产。
46、(3)本发明人意料地发现,补充加入tbuxphos配体等物质,产物难以分离,增加了配体和底物的分离难度;本发明在不特殊引入三苯基膦或xphos等配体物质,能达到高收率、高质量的开环化合物;本发明开环反应收率达到73%,能很好避免生成其他位置开环杂质。
47、(4)本发明人还意料地发现,开环反应加入cux类催化剂,转化率和反应速率均明显提升,不加入cux类催化剂开环反应的反应速率明显降低,转化率不足。
- 还没有人留言评论。精彩留言会获得点赞!