一种甾体吲哚衍生物及其制备方法和应用
本发明属于药物合成,具体涉及一种甾体吲哚衍生物及其制备方法和应用。
背景技术:
1、因致病微生物所导致的感染性疾病一直威胁着人类健康。尽管自20世纪以来,随着公共及个人卫生条件的改善,尤其是抗感染药物的使用,极大的减少了因感染而导致的发病和死亡,但随着抗生素的使用,耐药菌的出现仍然是一个不可忽视的问题。随着细菌耐药性的快速产生于增长以及传播速度的加快,微生物感染所导致的死亡已经居于世界前列,如何应对日益加剧的微生物感染所导致的疾病依然是一个亟需解决的问题。开发新型的抑菌药物仍然是应对细菌感染的重要手段。
技术实现思路
1、有鉴于此,本发明的目的在于提供一种甾体吲哚衍生物及其制备方法和应用,本发明提供的甾体吲哚衍生物具有良好的抑菌(铜绿假单胞菌、茄科雷尔氏菌、耐甲氧西林金黄色葡萄球菌和金黄色葡萄球菌)活性,具有进一步研究开发的前景。
2、为了实现以上目的,本发明提供了一种技术方案:
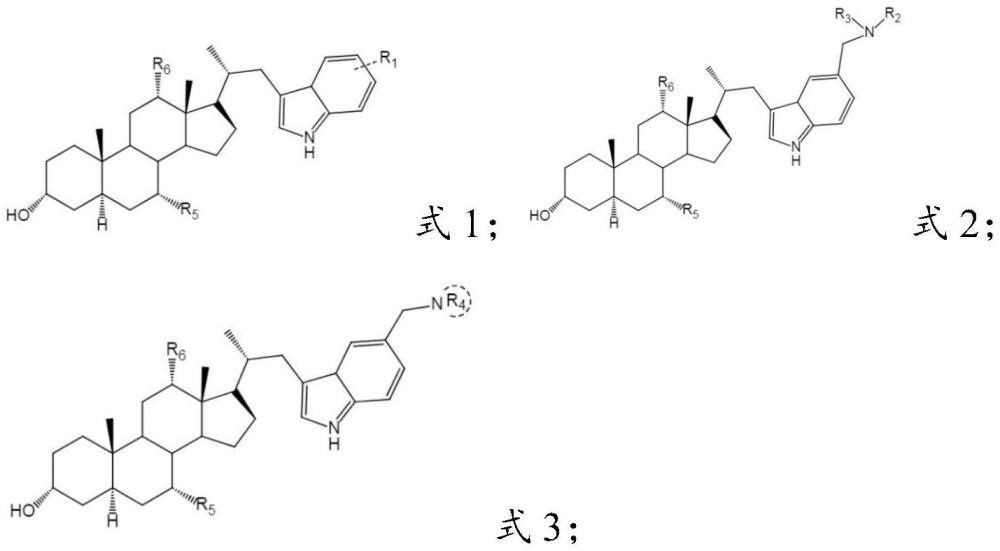
3、本发明提供了一种甾体吲哚衍生物,具有式1、式2或式3所示结构:
4、
5、r1为氢、羟基、甲基、甲氧基、醛基、酯基、卤素或羟甲基;
6、r2和r3独立地为c1-c6烷基、羟基取代c1-c6烷基、芳基或取代芳基;
7、r4为c4-c6环烷烃基、羟基取代c4-c6环烷烃基、哌嗪基、吗啉基或硫代吗啉基;
8、r5和r6独立地为氢或羟基。
9、优选地,所述c1-c6烷基为支链或直链结构;所述羟基取代c1-c6烷基为支链或直链结构。
10、优选地,所述芳基或取代芳基中碳原子数为6~10;所述芳基为苯基、萘基或c8芳环基团。
11、本发明还提供了所述的甾体吲哚衍生物的制备方法,当所述甾体吲哚衍生物具有式1所示结构时,制备方法包括以下步骤:
12、将具有式a结构的化合物a和具有式b结构的化合物b在酸性催化剂条件下进行亲核加成反应,得到具有式1所示结构的化合物1;
13、
14、当所述甾体吲哚衍生物具有式2或式3所示结构时,制备方法包括以下步骤:
15、将具有式c所示结构的化合物c和还原剂混合于四氢呋喃,进行还原反应,得到具有式d所示结构的化合物d;
16、
17、
18、将化合物d和二氧化锰混合于二氯甲烷,进行氧化反应,得到具有式e所示结构的化合物e;
19、
20、将化合物e、胺类化合物和三乙酰基硼氢化钠混合于二氯甲烷,进行胺化还原反应,得到具有式2或3所示结构的化合物2或3;
21、所述胺类化合物具有式g或式f所示结构:
22、
23、优选地,所述化合物a和化合物b的摩尔比为1:1~1:2;所述亲核加成反应的温度为40~100℃,时间为3~16h。
24、优选地,所述还原剂包括四氢铝锂、硼氢化钠和硼烷中的一种或几种;所述化合物c和还原剂的摩尔比为1:0.5~1:2;
25、所述还原反应的温度为10~30℃,时间为4~48h。
26、优选地,所述化合物d和二氧化锰的摩尔比为1:1~1:10;所述氧化反应的温度为10~30℃,时间为3~12h。
27、优选地,所述化合物e、胺类化合物和三乙酰基硼氢化钠的摩尔比为1:1~1:2;所述胺化还原反应的温度为10~30℃,时间为2~24h。
28、本发明还提供了上述所述的甾体吲哚衍生物在制备抑菌药物中的应用,所述应用时,抑制的细菌为铜绿假单胞菌、茄科雷尔氏菌、耐甲氧西林金黄色葡萄球菌和金黄色葡萄球菌中的一种或几种。
29、优选地,所述抑菌药物包括药学上可接受的赋形剂或载体以及所述的甾体吲哚衍生物;所述甾体吲哚衍生物在抑菌药物中的质量百分含量为10%~100%。
30、本发明提供了一种甾体吲哚衍生物,具有式1、式2或式3所示结构:r1为氢、羟基、甲基、甲氧基、醛基、酯基、卤素或羟甲基;r2和r3独立地为c1-c6烷基、羟基取代c1-c6烷基、芳基、取代芳基;r4为c4-c6环烷烃基、羟基取代c4-c6环烷烃基、哌嗪基、吗啉基或硫代吗啉基;r5和r6独立地为氢或羟基。本发明提供的甾体吲哚衍生物具有良好的抑菌(铜绿假单胞菌、茄科雷尔氏菌、耐甲氧西林金黄色葡萄球菌和金黄色葡萄球菌)活性,具有进一步研究开发的前景。
技术特征:
1.一种甾体吲哚衍生物,其特征在于,具有式1、式2或式3所示结构:
2.根据权利要求1所述的甾体吲哚衍生物,其特征在于,所述c1-c6烷基为支链或直链结构;所述羟基取代c1-c6烷基为支链或直链结构。
3.根据权利要求1所述的甾体吲哚衍生物,其特征在于,所述芳基或取代芳基中碳原子数为6~10;所述芳基为苯基、萘基或c8芳环基团。
4.权利要求1~3任一项所述的甾体吲哚衍生物的制备方法,其特征在于,当所述甾体吲哚衍生物具有式1所示结构时,制备方法包括以下步骤:
5.根据权利要求4所述的制备方法,其特征在于,所述化合物a和化合物b的摩尔比为1:1~1:2;所述亲核加成反应的温度为40~100℃,时间为3~16h。
6.根据权利要求4所述的制备方法,其特征在于,所述还原剂包括四氢铝锂、硼氢化钠和硼烷中的一种或几种;所述化合物c和还原剂的摩尔比为1:0.5~1:2;
7.根据权利要求4所述的制备方法,其特征在于,所述化合物d和二氧化锰的摩尔比为1:1~1:10;所述氧化反应的温度为10~30℃,时间为3~12h。
8.根据权利要求4所述的制备方法,其特征在于,所述化合物e、胺类化合物和三乙酰基硼氢化钠的摩尔比为1:1~1:2;所述胺化还原反应的温度为10~30℃,时间为2~24h。
9.权利要求1~3任一项所述的甾体吲哚衍生物在制备抑菌药物中的应用,所述应用时,抑制的细菌为铜绿假单胞菌、茄科雷尔氏菌、耐甲氧西林金黄色葡萄球菌和金黄色葡萄球菌中的一种或几种。
10.根据权利要求9所述的应用,其特征在于,所述抑菌药物包括药学上可接受的赋形剂或载体以及权利要求1~3任一项所述的甾体吲哚衍生物;所述甾体吲哚衍生物在抑菌药物中的质量百分含量为10%~100%。
技术总结
本发明属于药物合成技术领域,具体涉及一种甾体吲哚衍生物及其制备方法和应用。本发明提供了一种甾体吲哚衍生物,具有式1、式2或式3所示结构:R1为氢、羟基、甲基、甲氧基、醛基、酯基、卤素或羟甲基;R2和R3独立地为C1‑C6烷基、羟基取代C1‑C6烷基、芳基或取代芳基;R4为C4‑C6环烷烃基、羟基取代C4‑C6环烷烃基、哌嗪基、吗啉基或硫代吗啉基;R5和R6独立地为氢或羟基。本发明提供的甾体吲哚衍生物具有良好的抑菌(铜绿假单胞菌、茄科雷尔氏菌、耐甲氧西林金黄色葡萄球菌和金黄色葡萄球菌)活性,具有进一步研究开发的前景。
技术研发人员:李健,霍海波
受保护的技术使用者:长治学院
技术研发日:
技术公布日:2024/5/16
- 还没有人留言评论。精彩留言会获得点赞!