一种氮掺杂碳量子点的功能化无机荧光微球及其制备方法与流程
本发明属于功能化无机荧光微球制备方法技术领域,具体涉及一种氮掺杂碳量子点的功能化无机荧光微球及其制备方法。
背景技术:
碳量子点(CDs)作为一种新型的荧光探针,相比传统的半导体量子点和有机染料,具备宽而连续的激发光谱、窄而对称的发射光谱,良好的生物相容性和低毒性,在生物医学、标记、重金属检测等领域有良好的应用前景。但CDs荧光量子产率相对较低,研究表明在碳量子点表面进行氮掺杂可钝化表面缺陷且促进高产辐射复合,从而改善其荧光性能及水溶性。因而本文以白菜为碳源、多乙烯多胺为氮源,制备了绿色无毒的氮掺杂碳量子点(NCDs)。据文献报道,无机材料三聚氰胺甲醛具有耐热、耐老化及易溶于诸多有机溶剂等优良性能,其末端含有-NH2、-NH、-OH等基团,可与生物基团键合实现进一步的应用。脲醛树脂是亲水性的材料,具有机械强度高、热稳定性好、抗溶剂溶解、抗生物分解作用强,及制备容易等优点。二氧化硅具有物理刚性、化学稳定性、无毒性、胶体稳定性、高生物相容性及表面易功能化的特性,尤其是表面巯基化的二氧化硅微球,其活性巯基可以用来吸附重金属离子和蛋白质。由此,本文将NCDs作为荧光探针,分别以无机材料三聚氰胺甲醛、脲醛树脂和二氧化硅作为载体,用于功能化无机荧光微球的制备。
三聚氰胺甲醛微球含有丰富的氨基、亚氨基和羟基基团,可以供应大量的吸附活性点,用来吸附重金属离子Pb2+,但是其吸附活性点在生物上的应用还很少(Colloids and Surfaces A:Physicochem.Eng.Aspects,2015,482,491-499 and Chemical Engineering Journal,2016,288,745-757)。脲醛树脂微球的制备方法报道很多,但忽略了在其表面引入功能基团,不便后续生物学等方面的研究(Materials Letters,2015,145,27-29 and Journal of Industrial and Engineering Chemistry,201,218,919-925)。单分散大孔二氧化硅微球和巯基化二氧化硅微球都可以用于蛋白质的分离,但大孔二氧化硅微球的制备过程繁杂,巯基化二氧化硅微球易于制备(Journal of Chromatography A,2016,1471,138-144 and Journal of Applied Polymer Science,2015,132(20),113-114)。而本文制备的发蓝色荧光的三聚氰胺甲醛荧光微球,可以应用于流式细胞仪(Journal of Photochemistry and Photobiology B:Biology,2013,129,125-134)和荧光免疫检测(Science of the Total Environment,2017,583,222-227)方面;脲醛树脂荧光微球和二氧化硅荧光纳米球分别经15分钟和3个小时就可以制备得到,不仅实验操作时间短,还在其表面分别引入了硅羟基和巯基。本文获得的三种功能化无机荧光微球单分散性良好、功能基含量丰富,易与生物分子结合,体现了良好的生物相容性和低毒性,在生物、医学领域具有很好的应用前景。
技术实现要素:
本发明的目的是提供一种具有良好的单分散性,荧光性能好且稳定的氮掺杂碳量子点的功能化无机荧光微球的制备方法。
一种氮掺杂碳量子点的功能化无机荧光微球的制备方法,包括以下步骤:
(1)氮掺杂碳量子点NCDs的制备
量取35~50mL白菜汁,并转移至反应釜中,置于烘箱中180~200℃加热5~7h;反应结束后,自然冷却至室温,采用孔径0.22μm的微孔滤膜过滤产物,将滤液在12000r/min转速下离心,去除大颗粒杂质;在纯化后的溶液中分别加入一定量氨水,尿素,二乙烯三胺,多乙烯多胺,继续在180~200℃的烘箱中反应5~7h;反应后将反应釜自然冷却至室温,得到棕色溶液,采用孔径0.22μm的微孔滤膜过滤,将滤液在12000r/min转速下离心,去除大颗粒杂质;并用1000分子量的透析袋透析所得滤液,最后用冷冻干燥法处理,得到纯净的氮掺杂碳量子点NCDs,其为疏松多孔、海绵状的固体;
(2)三聚氰胺甲醛荧光微球的制备
将摩尔比为3.5~3.0的甲醛和三聚氰胺加入到100mL三口烧瓶中,机械搅拌混合均匀,水浴加热到60~65℃,反应30~35min后得到均匀的预聚物羟甲基三聚氰胺;
量取42~48mL蒸馏水和0.2~0.5mL步骤(1)中所述的纯净的氮掺杂碳量子点NCDs,,称取1.3g聚乙烯吡咯烷酮,混合后加入到250mL三口烧瓶中,用冰乙酸调节溶液的pH为4.5~4.6,机械搅拌下水浴加热到60~65℃,将所述的预聚物羟甲基三聚氰胺迅速加入其中,使用冰乙酸迅速微调pH到4.5~4.6,当出现白色浑浊后,每隔10min在显微镜下观察微球的形貌,根据生成球的情况调整反应时间,反应15~30min后加入大量冰水停止反应;依次用蒸馏水、无水乙醇反复洗涤后真空干燥,即得三聚氰胺甲醛荧光微球;
(3)羟基脲醛树脂荧光微球的制备
在塑料杯中加入42~45mL蒸馏水、4.2~4.5gNaCl、0.8~1g尿素和0.2~0.5mL步骤(1)中所述的纯净的氮掺杂碳量子点NCDs,,待NaCl和尿素完全溶解后用硝酸将混合液的pH值调为1.7~2.0,然后加入1.3~1.8mL甲醛溶液,在室温下放置2~15min,期间不断用显微镜监测微球的形貌;当微球粒径达到均一时,加入100~150mL蒸馏水结束反应,静置沉降后抽滤,滤饼再分别用甲醇、丙酮抽洗直至上清液无荧光为止,50~60℃真空干燥,即得到脲醛树脂荧光微球;
将2g脲醛树脂荧光微球置于三口烧瓶中,加入乙醇80mL,蒸馏水16mL,四乙氧基硅烷0.5mL,氨水2mL,在室温下,剧烈搅拌3h,然后再用乙醇和蒸馏水反复洗涤,弃去上清液,50~60℃真空干燥,即得到羟基脲醛树脂荧光微球;
(4)巯基化二氧化硅荧光纳米球的制备
按质量比1:4:8量取氨水、蒸馏水、无水乙醇加入到反应瓶中,混和均匀,再加入3.5~4.5mL正硅酸乙酯和0.1~0.5mL步骤(1)中所述的纯净的氮掺杂碳量子点NCDs,,室温下磁力搅拌3~3.5h;将产物离心分离,用水和无水乙醇超声清洗至上清液无荧光;
50~60℃真空干燥,即得到二氧化硅荧光纳米球;将0.3~0.4g的SiO2荧光纳米球和0.2~0.3mL的γ-巯丙基三甲氧基硅烷KH590加入到三口烧瓶中,并加入无水甲苯,使无水甲苯:KH590为1:75,在回流条件下反应24h,即得到巯基化二氧化硅荧光纳米球。
前述步骤(1)中用1000分子量的透析袋透析所得溶液的时间为2d,条件为每6h换一次水。
前述步骤(1)中用冷冻干燥法处理的具体工艺为:首先将氮掺杂碳量子点溶液冻结成固态,然后在-55℃下冷冻干燥24~48h。
前述步骤(2)中真空干燥的温度为50~60℃,干燥时间为12~24小时。
一种氮掺杂碳量子点的功能化无机荧光微球,采用前述任意一种氮掺杂碳量子点的功能化无机荧光微球的制备方法所制备。
本发明的有益之处在于:本发明中的一种氮掺杂碳量子点的功能化无机荧光微球的制备方法,采用简单易行的水热法制备了绿色无毒的白菜碳量子点,氮掺杂后呈现更好的荧光性能;将氮掺杂碳量子点作为荧光探针,应用于荧光微球制备方面,获得了表面氨基化的三聚氰胺甲醛荧光微球、羟基脲醛树脂荧光微球和巯基二氧化硅纳米荧光球。这些发蓝色荧光的荧光微球单分散性良好、功能基含量丰富,可以与生物分子结合,在免疫分析、高通量药物筛选、药物载体、固定化酶、细菌和病毒诊断、细胞因子鉴定以及单细胞分析等方面具有良好的应用前景。
附图说明
图1是本发明的实施例中氮掺杂碳量子点的制备的合成示意图;
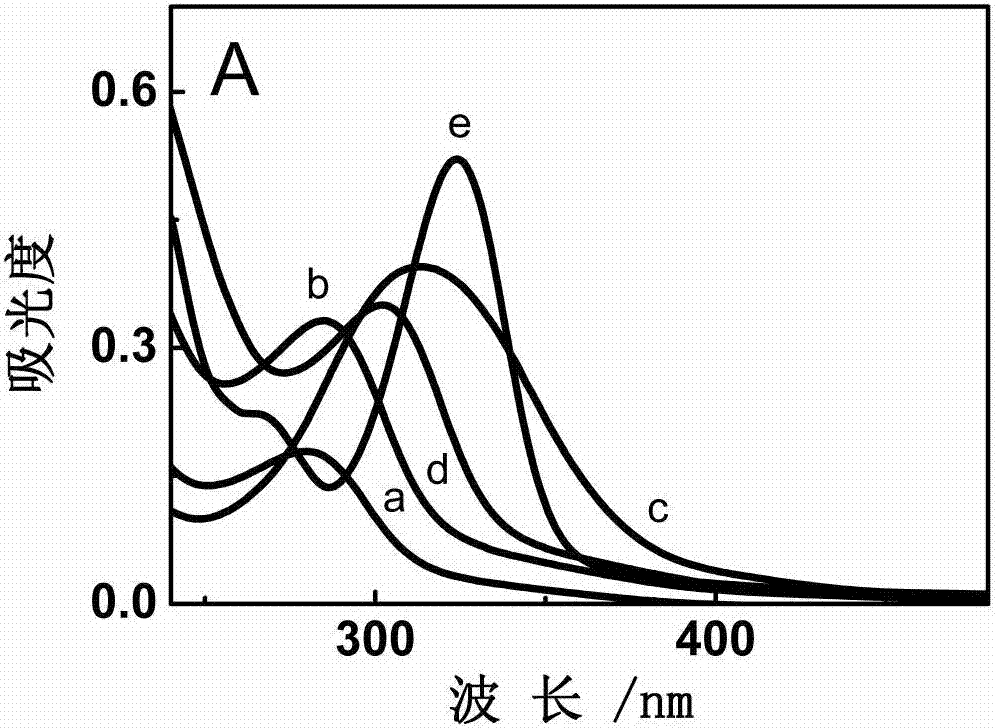
图2是本发明的实施例中氮掺杂碳量子点的紫外-可见吸收光谱;
图3是本发明的实施例中氮掺杂碳量子点的荧光发射光谱;
图4是本发明的实施例中氮掺杂碳量子点的红外谱图;
图5是本发明的实施例中具有氨基的三聚氰胺甲醛荧光微球的制备过程图;
图6是本发明的实施例中三聚氰胺甲醛微球、三聚氰胺甲醛荧光微球的扫描电镜图和荧光照片;
图7是本发明的实施例中的三聚氰胺甲醛微球、三聚氰胺甲醛荧光微球的红外谱图;
图8是本发明的实施例中制备的三聚氰胺甲醛荧光微球和氮掺杂碳量子点的荧光光谱图。
图9是本发明的实施例中羟基脲醛树脂荧光微球的制备过程图;
图10是本发明的实施例中羟基脲醛树脂荧光微球、脲醛树脂荧光微球的红外谱图;
图11是本发明的实施例中脲醛树脂荧光微球、羟基脲醛树脂荧光微球的扫描电镜图和荧光照片;
图12是本发明的实施例中的羟基脲醛树脂荧光微球和氮掺杂碳量子点的荧光光谱图;
图13是本发明的实施例中的二氧化硅荧光纳米球的制备过程图;
图14是本发明的实施例中二氧化硅荧光纳米球、巯基二氧化硅荧光纳米球的红外谱图;
图15是本发明的实施例中巯基化二氧化硅荧光纳米球的扫描电镜图、投射电镜图和荧光照片;
图16是本发明的实施例中的巯基化二氧化硅荧光纳米球和氮掺杂碳量子点的荧光光谱图。
具体实施方式
氮掺杂碳量子点的功能化无机荧光微球的制备方法,包括以下步骤:
(1)氮掺杂碳量子点(NCDs)的制备
量取35mL白菜汁并转移至反应釜中,置于烘箱中200℃加热5h;反应结束后,自然冷却至室温,采用孔径0.22μm的微孔滤膜过滤产物,将滤液在12000r/min转速下离心,去除大颗粒杂质;在纯化后的溶液中分别加入一定量氨水,尿素,二乙烯三胺,多乙烯多胺,继续在200℃的烘箱中反应5h。反应后将反应釜自然冷却至室温,得到棕色溶液,采用孔径0.22μm的微孔滤膜过滤产物,将滤液在12000r/min转速下离心,去除大颗粒杂质;并用1000分子量的透析袋透析所得溶液的时间为2d,条件为每6h换一次水;最后用冷冻干燥法处理得到纯净的氮掺杂碳量子点,呈现出疏松多孔、海绵状的固体,合成过程如图1所示。
对氮掺杂碳量子点的固体进行表征,结果如下:
参考图2为在相同浓度下在CDs中分别加入尿素(b),氨水(c),二乙烯三胺(d),多乙烯多胺(e)的实验组和CDs(a)对照组的紫外-可见吸收光谱。由图可知,实验组较对照组的最大吸收峰由280nm红移至350nm,这是因为经钝化剂尿素、氨水、二乙烯三胺、多乙烯多胺修饰后,其吸收波长会相应增加。
图3为在相同浓度下在CDs中分别加入尿素(b),氨水(c),二乙烯三胺(d),多乙烯多胺(e)的实验组和CDs(a)对照组的荧光发射光谱,激发波长为相应紫外的最大吸收波长。由图可知,氮掺杂碳量子点荧光强度逐渐增强,且发射波长发生了红移,这是由于有机溶剂钝化,使CDs表面产生能量势阱,且随着氨基含量的增加而增加,从而导致NCDs的荧光增强。CDs和NCDs溶液在自然光下均呈现淡黄色透明状;在365nm紫外灯激发下,NCDs比CDs有更高的荧光强度且呈现亮蓝色。这与发射波长433nm~470nm是一致的,进一步揭示了CDs和NCDs具有强烈的蓝色荧光。选多乙烯多胺氮掺杂碳量子点为荧光探针应用于荧光微球制备方面,以0.1mol/L H2SO4,54%的硫酸奎宁为标准参照物,测得CDs的荧光量子产率为26.2%,NCDs的荧光量子产率为53.3%,实验结果说明进行多乙烯多胺氮掺的碳量子点的荧光量子产率高。
参照图4为实验组NCDs和对照组CDs和的红外谱图。对照组CDs在3394cm-1处为羟基特征吸收峰,1638cm-1为C=O伸缩振动峰,1386cm-1和1097cm-1处为-OH和C-H的弯曲振动峰;实验组NCDs在3455cm-1处为O-H的伸缩振动或在多乙烯多胺条件下水热产生的N-H伸缩振动,在2932cm-1、1712cm-1、1397cm-1和1184cm-1处的吸收分别对应C-H、C=O、C-N和C-O-C伸缩振动。结果表明,所制得的NCDs表面富含-NH2、-C=O、-OH等含氧官能团,使NCDs的亲水性和稳定性得到提高,且在水溶液中表现出电负性。
(2)三聚氰胺甲醛荧光微球的制备
将1.4g三聚氰胺和2.7mL甲醛加入到100mL三口烧瓶中,机械搅拌混合均匀;水浴加热到60℃,反应30min后得到均匀的预聚物羟甲基三聚氰胺;
量取45mL蒸馏水和0.5mL的氮掺杂碳量子点的固体粉末,称取1.3g聚乙烯吡咯烷酮做分散剂,混合加入到250mL三口烧瓶中,用冰乙酸调节溶液的pH为4.5~4.6,机械搅拌下水浴加热到60℃;将预聚物羟甲基三聚氰胺迅速全部加入其中,使用冰乙酸迅速微调pH到4.5~4.6,当出现白色浑浊后,每隔10min在显微镜下观察微球的形貌,根据生成球的情况调整反应时间;反应30min后加入大量冰水停止反应,依次用蒸馏水、无水乙醇反复洗涤后,50℃真空干燥,即得三聚氰胺甲醛荧光微球的制备,制备过程如图5所示;
对三聚氰胺甲醛荧光微球进行表征,结果如下:
参考图6,A、B和C分别为三聚氰胺甲醛微球、三聚氰胺甲醛荧光微球的扫描电镜图和荧光照片。由图可知,三聚氰胺甲醛荧光微球单分散性良好,NCDs在微球中分布均匀,使得微球发光效率高且很稳定,这是NCDs与三聚氰胺甲醛微球形成的作用力密切相关。首先三聚氰胺经羟甲基化形成预聚物,然后通过羟甲基之间或羟甲基与氨基、亚胺基之间发生缩聚反应形成醚键或亚甲基,同时NCDs中的羟基、氨基和羰基基团与预聚物中的亚氨基和羟甲基形成牢固的氢键,最后逐渐成核与长大生成交联网状的三聚氰胺甲醛荧光微球,致使NCDs不易从三聚氰胺甲醛荧光微球中泄露出来。
参考图7,a为三聚氰胺甲醛微球的红外谱图,3410cm-1处较强的吸收带为N-H伸缩振动峰,1552cm-1与1492cm-1处与三嗪环上C=N伸缩振动及N-H剪切弯曲振动峰,在2940cm-1和1348cm-1处分别为C-H和环内C-N的伸缩振动峰,1158cm-1和1004cm-1处是醚键C-O-C和醇类的C-O伸缩振动峰,815cm-1处是三聚氰胺骨架的面外弯曲振动吸收峰;b是三聚氰胺甲醛荧光微球的红外谱图,其峰型和官能团的归属基本与三聚氰胺甲醛微球一致,这是由于NCDs在三聚氰胺甲醛荧光微球中含量极少,红外图谱不能表征出NCDs的特征峰。
参考图8,三聚氰胺甲醛荧光微球的最大发射波长(b)相对于NCDs(a)发生了一定的移动,这是由于当碳量子点包埋在微球中,其荧光光谱会发生一定的变化。
(3)羟基脲醛树脂荧光微球的制备
在塑料杯中加入42mL蒸馏水,4.2g NaCl、1g尿素和0.4mL氮掺杂碳量子点,待NaCl和尿素完全溶解后用硝酸将混合液的pH值调为1.7,然后加入1.5mL甲醛溶液,在室温下放置一定时间,在显微镜下观察有微球产生;加入125mL蒸馏水,静置沉降后抽滤,滤饼再分别用甲醇、丙酮抽洗,直至上清液无荧光为止,60℃真空干燥24h,即得到脲醛树脂荧光微球;
将2g脲醛树脂荧光微球置于250ml三口瓶中,加入乙醇80ml,蒸馏水16ml,四乙氧基硅烷0.5ml,氨水2ml;室温下,剧烈搅拌3h;然后在下用乙醇和蒸馏水反复洗涤,弃去清液,60℃真空干燥24h,即得到羟基脲醛树脂荧光微球,制备过程如图9所示;
对羟基脲醛树脂荧光微球进行表征,结果如下:
参考图10,a是羟基脲醛树脂荧光微球的红外谱图,b是脲醛树脂荧光微球的红外谱图;图a中3348cm-1处的峰归属于-NH-,1642cm-1是-C=O官能团的峰,因此可以确定有-NH-C=O-存在,-NH-C=O-是脲醛树脂中主要的官能团;1090cm-1附近归属于Si-O-Si,表明脲醛树脂荧光微球表面包覆了一层二氧化硅。
参考图11,A、B和C分别为脲醛树脂荧光微球、羟基脲醛树脂荧光微球的扫描电镜图和荧光照片;由图可知,脲醛树脂荧光微球单分散性良好,NCDs在微球中分布均匀,使得微球发光效率高且很稳定,这与NCDs与脲醛树脂微球形成的氢键作用力密切相关。
参考图12,脲醛树脂荧光微球的最大发射波长(b)相对于NCDs(a)发生了一定的移动。这是由于当碳量子点包埋在微球中,其荧光光谱会发生一定的变化。
(5)巯基化二氧化硅荧光纳米球的制备
在28mL蒸馏水,56mL乙醇,7mL氨水的混合溶液中加入4.2mL正硅酸乙酯和0.4mL氮掺杂碳量子点,在室温下磁力搅拌3个小时;将产物离心分离,用水和无水乙醇超声清洗至上清液无荧光;50~60℃下真空干燥,即得到二氧化硅荧光纳米球,制备过程如图13所示。
将0.35g SiO2荧光纳米球和0.25mLγ-巯丙基三甲氧基硅烷(KH590)加入到三口烧瓶中,并加入18mL无水甲苯,在回流条件下反应24h。
对巯基化二氧化硅荧光纳米球进行表征,结果如下:
参考图14,a是二氧化硅荧光纳米球的红外谱图,b是巯基二氧化硅荧光纳米球的红外谱图。图a、b中3429cm-1处的峰归属于Si-OH;图b中2924cm-1和2851cm-1处有新峰出现,为巯基硅烷偶联剂上-CH2-的特征峰,表明有巯基引入。
参考图15,A、B和C分别为巯基化二氧化硅荧光纳米球的扫描电镜图、投射电镜图和荧光照片。由图可知,巯基化二氧化硅荧光纳米球的单分散性良好,NCDs在微球中分布均匀,使得微球发光效率高且很稳定。
参考图16,巯基化二氧化硅荧光纳米球的最大发射波长(a)相对于NCDs(b)发生了一定的移动。这是由于当碳量子点包埋在微球中,其荧光光谱会发生一定的变化。
应当理解,以上所描述的具体实施例仅用于解释发明,并不用于限定本发明。由发明的精神所引伸出的显而易见的变化或变动仍处于本发明的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!