一种氮磷掺杂石墨烯载钯催化剂及其制备方法与流程
本发明涉及一种石墨烯催化剂,具体涉及一种氮磷掺杂石墨烯载钯催化剂及其制备方法,属于燃料电池纳米电催化剂制备领域。
背景技术
随着社会的不断发展与进步,环境污染和能源危机成为了我们人类社会进步必须要突破的难题。燃料电池因为它拥有着干净,安全,低噪音,多样化的燃料,较高的转换效率(40~70%)的优点,迅速的占据人们的视线,选择优秀的体系,高性能的催化剂成为了现在专家学者研究的主要方向。
在甲醇燃料电池中pt基催化剂被认为是最好的催化剂,但是pt成本高,资源有限,并且易受co中毒,从而限制了pt基催化剂的大规模使用。pd被认为是较好的替代物,具有成本低、抗co中毒性能更佳等优点,因而是近些年研究的热点。催化剂性能很大程度上受载体材料的影响。一般理想的电催化剂载体材料应同时具备高比表面积、高导电性、合适的孔结构、优异的耐腐蚀性以及合适的表面功能团[3-6]。作为一种新型的二维碳材料,石墨烯材料具有优异的电子传导能力、超大比表面积(理论比表面积为2620m2·g-1)及特殊的力学、量子和电学性质,这使它在催化剂载体方面有着非常好的应用前景[7,8]。但是目前石墨烯作为载体载pd催化剂的制备,一种方法是采用微机械剥离高定向热解石墨(hopg)法和化学气相沉积法(cvd)等制备的石墨烯作为载体。这类石墨烯晶体结构完整,电子传导能力及力学性质都很优异,但是表面并不活泼,不容易掺杂金属粒子。另一种是采用hummers法液相氧化合成氧化石墨烯(go),然后用化学一步还原制得石墨烯负载钯催化剂,这也是目前更多采用的合成方法。可是这种制备也存在问题,一是石墨烯表面含有大量氧物种,导致石墨烯导电性能达不到理想值。而通过杂原子掺杂可以对石墨烯表面进行改性,可以调变石墨烯表面的电子性质。比如b-n掺杂石墨烯,n原子掺杂可以调变c原子的自旋密度和电荷分布,而b原子的共同参与,加强了这种不对称的自旋密度,也促进了石墨烯平面结构中的电子流动性,即加强了石墨烯的导电性。
技术实现要素:
本发明的目的在于提供一种电催化性能较为优异的氮磷掺杂石墨烯载钯催化剂(pd/n-p-g及pd/p-n-g),该催化剂通过元素掺杂表面修饰技术,在石墨烯表面引入不同的活性位,提供强导电性和有利于附着金属pd的载体作用。
本发明解决其技术问题所采用的技术方案是:
一种氮磷掺杂石墨烯载钯催化剂,该催化剂中,含有p3.12±0.2%,n7.41±0.5%,pd2.93±0.5%,c72.65±5%,余量为o,以上均为原子比。
以pd/n-p-g为例,该催化剂由如下方法制得:以hummers合成法制得氧化石墨烯,高压反应釜中加入二氰二胺溶液,过滤干燥后,氩气保护下400-550℃焙烧进行高温热解;再高压反应釜加入磷酸溶液,抽滤干燥后氩气保护下500-650℃焙烧进行高温热解得到氮磷掺杂石墨烯;再采用硼氢化钾法负载pd金属,得到氮磷掺杂石墨烯载钯催化剂。本发明是以hummers合成法制得氧化石墨烯,分别以二氰二胺和磷酸为n源和p源,再分步进行高压釜中浸渍再惰性保护高温热解进行掺杂。再将产品经硼氢化钾法负载上pd金属,得到氮磷掺杂石墨烯载钯催化剂。
一种所述的氮磷掺杂石墨烯载钯催化剂的制备方法,该方法包括如下步骤:以hummers合成法制得氧化石墨烯,先以二氰二胺为n源,在高压釜中浸渍并在惰性保护下400-550℃高温热解进行掺杂,再以磷酸为p源,在高压釜中浸渍并在惰性保护下500-650℃高温热解进行掺杂;最后将其经硼氢化钾法负载上pd金属,得到氮磷掺杂石墨烯载钯催化剂。
作为优选,该方法包括如下步骤:
①n-go的制备
质量浓度为20-50%的二氰二胺溶液与氧化石墨烯一起用超声波破碎仪超声30-40min至混合均匀呈油墨状,将所得溶液转入聚四氟乙烯内衬的高压反应釜中,150-250℃反应6-8h后自然冷却,抽滤干燥;所得固体转至石英舟,置于节能管式炉中,通氩气400-550℃焙烧进行高温热解2-4h,得到n-go;
②n-p-go的制备
取n-go与质量浓度为10-50%的磷酸溶液混合,超声均匀后,转移至高压反应釜,130-230℃下反应5-6小时后取出,进行抽滤,将所得固体转至石英舟置于节能管式炉中,氩气保护下500-650℃下焙烧2-4h,反应结束后,冷却,得到n-p-go载体;
③pd/n-p-g的制备
将上一步制得的n-p-go载体加入适量去离子水,超声15-20min均匀至油墨状,再加入pdcl2溶液;超声完成后,用氨水溶液调节ph值至弱碱性,在搅拌下逐滴加入kbh4溶液,50-55℃冷凝回流反应4-6h,趁热过滤,洗涤,70±5℃烘干,研磨后即得pd/n-p-g催化剂。
作为优选,所述二氰二胺溶液的质量浓度为28-32%,磷酸的质量浓度为28-32%。
一种所述的氮磷掺杂石墨烯载钯催化剂的制备方法,该方法包括如下步骤:以hummers合成法制得氧化石墨烯,先以磷酸为p源,在高压釜中浸渍并在惰性保护下500-650℃高温热解进行掺杂;再以二氰二胺为n源,在高压釜中浸渍并在惰性保护下400-550℃高温热解进行掺杂;最后将其经硼氢化钾法负载上pd金属,得到氮磷掺杂石墨烯载钯催化剂。
作为优选,该方法包括如下步骤:
①p-go的制备
氧化石墨烯与质量浓度为10-50%的磷酸溶液混合,超声至油墨状,倒入高压反应釜中,130-230℃下干燥箱反应5-6h,冷却减压抽滤干燥,将所得固体转至石英舟置于节能管式炉中,氩气保护下500-650℃下焙烧2-4h,得到p-go;
②p-n-go的制备
质量浓度为20-50%的二氰二胺溶液与p-go一起用超声波破碎仪超声至油墨状,将所得混合物转入聚四氟乙烯内衬的高压反应釜中,150-250℃反应6-8h后自然冷却,抽滤干燥,所得固体转至石英舟,置于节能管式炉中,通氩气400-550℃焙烧进行高温热解2-4h,得到p-n-go;
③pd/p-n-g的制备
将上一步制得的p-n-go载体加入适量去离子水,超声15-20min均匀至油墨状,再加入pdcl2溶液;超声完成后,用氨水溶液调节ph值至弱碱性,在搅拌下逐滴加入kbh4溶液,50-55℃冷凝回流反应4-6h,趁热过滤,洗涤,70±5℃烘干,研磨后即得pd/p-n-g催化剂。
作为优选,所述二氰二胺溶液的质量浓度为28-32%,磷酸的质量浓度为28-32%。
本发明将氮磷掺杂石墨烯载钯催化剂,与单元素掺杂石墨烯(n掺杂和p掺杂)载钯催化剂,无掺杂石墨烯载钯催化剂进行cv(循环伏安特性曲线),lsv等电化学性能表征,将所得的表征图像进行对比,获得氮磷掺杂石墨烯最佳技术路线。从对比图像中可以读出这五种催化剂自身电催化性能的优劣,其中以本发明的氮磷掺杂石墨烯载钯催化剂性能最优(pd/n-p-g),最后将催化剂在纯氢氧化钾(koh)体系中所得的表征图像与在nch3oh:nkoh=1:1的溶液体系中的表征图像做对比,得出后者体系下,催化剂的电催化性能更佳。
附图说明
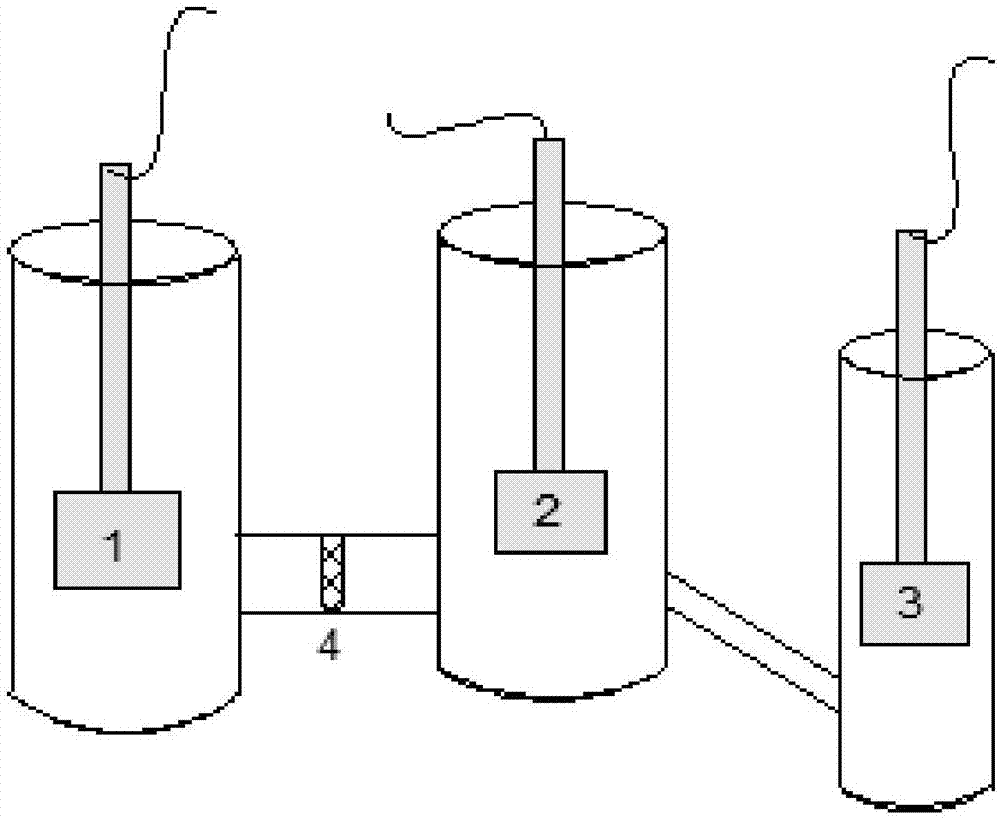
图1是用于催化剂的电化学性能表征的实验装置图;
图2是5种催化剂电极在1mol/lkoh+1mol/lch3oh的电解质溶液中的循环伏安曲线,扫描速度为50mv/s;
图3是5种电极在1mol/lkoh+1mol/lch3oh的电解质溶液中的线性扫描伏安曲线,扫描速度为2mv/s;
图4是5种催化剂电极在1mol/lkoh电解液中的循环伏安曲线谱图,扫描速度为20mv/s;
图5是5种催化剂电极在-0.2v恒电位下在1mol/lkoh+1mol/lch3oh的电解质溶液中的极化电流曲线;
图6是5种催化剂的xrd图;
图7是(a)pd/n-p-g,(b)pd/p-n-g,(c)pd/p-g和(d)pd/g催化剂的透射电镜tem图;
图8是(a)pd/n-p-g和pd/p-g催化剂的xps图,(b)pd/n-p-g催化剂的n1s谱峰图,(c)、(d)、(e)分别是pd/p-g,pd/n-p-g和pd/p-n-g的催化剂p2p谱峰图,(f)、(g)分别是pd/n-p-g和pd/p-n-g的催化剂pd3d谱峰图,(h)4种催化剂的pd谱峰对比图。
具体实施方式
下面通过具体实施例,对本发明的技术方案作进一步的具体说明。应当理解,本发明的实施并不局限于下面的实施例,对本发明所做的任何形式上的变通和/或改变都将落入本发明保护范围。
在本发明中,若非特指,所有的份、百分比均为重量单位,所采用的设备和原料等均可从市场购得或是本领域常用的。下述实施例中的方法,如无特别说明,均为本领域的常规方法。
主要仪器为:chi760d电化学分析仪,d8advance型x射线衍射仪器,日本jeol-2010透射电子显微镜,escalab250xi光电子能谱仪。
实施例:
一、pd/n-p-g催化剂的合成
1n-go的制备
称取21.429g的二氰二胺,加50ml去离子水溶解,再加入200mg氧化石墨烯,用超声波破碎仪超声30min至混合均匀呈油墨状。将所得溶液转入聚四氟乙烯内衬的高压反应釜中,180℃反应6-8h后自然冷却,减压抽滤干燥。所得固体转至石英舟,置于节能管式炉中,通氩气400-550℃焙烧进行高温热解2-4h,得到n-go。
2n-p-go的制备
称取100mg的n-go与50ml磷酸溶液(质量分数30%)混合,超声均匀后,转移至高压反应釜,150℃下反应5-6小时后取出,进行减压抽滤,将所得固体转至石英舟置于节能管式炉中,氩气保护下500-650℃下焙烧2-4h,反应结束后,冷却,得到n-p-go载体。
3pd/n-p-g的制备
将上一步制得的80mgn-p-go载体加入50ml去离子水,超声15min均匀至油墨状,再加入2.82ml的0.05mol/l的pdcl2溶液;超声完成后,用氨水溶液调节ph值至弱碱性,在搅拌下逐滴加入120ml的2.0mg/ml的kbh4溶液,50℃冷凝回流反应4-6h,趁热过滤,洗涤,70℃烘干,研磨后即得pd/n-p-g催化剂。
二、pd/p-n-g催化剂的合成
1p-go的制备
称取200mg氧化石墨烯,50ml磷酸溶液(30%)混合,超声至油墨状。倒入高压反应釜中,在150℃干燥箱反应5-6h,冷却减压抽滤干燥,将所得固体转至石英舟置于节能管式炉中,氩气保护下500-650℃下焙烧2-4h,得到p-go。
2p-n-go的制备
称取21.429g的二氰二胺,加50ml去离子水溶解,再加入100mg的p-go,用超声波破碎仪超声至油墨状。将所得混合物转入聚四氟乙烯内衬的高压反应釜中,180℃反应6-8h后自然冷却,抽滤干燥。所得固体转至石英舟,置于节能管式炉中,通氩气400-550℃焙烧进行高温热解2-4h,得到p-n-go。
3pd/p-n-g的制备
将上一步制得的80mgp-n-go载体加入50ml去离子水,超声15min均匀至油墨状,再加入2.82ml的0.05mol/l的pdcl2溶液;超声完成后,用氨水溶液调节ph值至弱碱性,在搅拌下逐滴加入120ml的2.0mg/ml的kbh4溶液,50℃冷凝回流反应4-6h,趁热过滤,洗涤,70℃烘干,研磨后即得pd/p-n-g催化剂。
三、催化剂的表征
1x射线衍射(xrd)
x射线衍射(xrd)测试:xrd测试在brukeraxsd8advance型x射线衍射仪上进行(cukα,λ=0.154056nm),工作电压40kv,电流40ma,扫描速度为2°/min,步宽0.04°。
2电极的制备
用分析天平称取5mg的pd/n-p-g催化剂,放入离心管中,用进样针移取nafion溶液和无水乙醇溶液(1:2)到离心管中,用超声清洗机将其超声至油墨状后,再用移液枪移取3μl于已磨好并干燥的电极中,(电极材料为经al2o3抛光后的玻璃碳,直径为4mm。)待到催化剂干燥后,就是实施例中要用的工作电极。
3实验装置的组装
采用三电极体系(如图1),1为铂片,2为工作电极,3为饱和甘汞电极,4为玻璃隔膜。参比电极:饱和甘汞电极,用于确定工作电极电位;辅助电极:铂电极,用于传导电流。使用cv、lsv和it研究催化剂对甲酸氧化的电催化性能。cv可以观察到催化剂的催化氧化的电流峰(扫描范围在-0.2v~0.8v之间,扫描速率为50mv/s和20mv/s);lsv可以确定催化剂的起始氧化电位;it可以测得甲酸电催化氧化电流随时间变化(恒电位为0.4v)。电解液为1mol/lch3oh+1mol/lkoh(以下均简称为电解液)。在电解液中通入20min得氮气以除去电解液中的溶解氧,方可进行电化学性能测试。
4电化学性能测试分析
4.1催化剂在1mol/lch3oh+1mol/lkoh的电化学性能
图2是不同载体负载pd纳米颗粒复合材料对甲醇的电催化氧化性能。正向扫描中,在-0.2v(vs.sce)附近出现的氧化峰来源于甲醇的氧化;反向扫描中,在-0.4v(vs.sce)附近出现的氧化峰可归属为含碳中间产物的二次氧化。通过正向扫描过程中氧化峰电流密度的大小反映出催化剂对甲醇氧化反应的电催化活性,氧化峰电流密度越大,则对甲醇氧化的电催化活性越强。从图中可以看出催化剂pd/n-p-g和pd/p-n-g峰电流密度分别为121.1ma·cm-2和92.6ma·cm-2,明显比pd/p-g(70.5ma·cm-2)、pd/n-g(42.6ma·cm-2)和pd/g(24.3ma·cm-2)的相应值高,对甲醇表现出更为良好的催化性能。这说明石墨烯经过n、p双掺杂后负载pd的催化剂性能明显优于单一的p掺杂或是n掺杂的电催化性能,可以得出n-p共掺杂石墨烯载体对pd的催化剂性能有更好的促进作用。
图3表示,在浓度为1mol/lch3oh+koh溶液中pd/p-n-g、pd/n-p-g、pd/p-g、pd/n-g、pd/g的线性扫描伏安曲线,扫描速度为2mv/s。图中可以明显地观察到:在电压处于-0.02v附近每个催化剂都产生了氧化峰,有掺杂p或n元素的石墨烯载钯催化剂比未掺杂p或n元素的石墨烯载钯催化剂pd/g具有更高的电流密度,双元素掺杂石墨烯载钯催化剂比单元素掺杂石墨烯载钯催化剂具有更高的电流密度,其中掺杂顺序pd/n-p-g比pd/p-n-g性能更好。这表明pd/n-p-g的催化剂在碱性介质中对甲醇氧化催化反应具有更好的性能。
4.2催化剂在1mol/lkoh体系中的电化学性能
为了研究催化剂的电化学性能,将其修饰的玻碳电极对甲醇的电催化氧化进行了测试,同时将不同催化剂进行比较。图4为pd/n-p-g、pd/p-n-g、pd/p-g、pd/n-g和pd/g在1mol·l-1koh溶液中的循环伏安曲线。从图中可以看出,在正扫过程中,大约在-0.7--0.4v(vs.sce)有一处宽峰,对应于氢的脱附峰;在回扫过程中,-0.45v的还原峰对应于pd(ⅱ)还原为pd(0),再往负电位偏移出现的肩峰则来源于氢的吸附峰。氢吸/脱附峰面积可以被用来定性评估催化剂的电化学活性表面积(ecsa),从图中可以明显看出,氢吸/脱附峰面积的大小顺序为:pd/n-p-g>pd/p-n-g≈pd/p-g>pd/n-g>pd/g。这说明在同样的负载量下,pd在pd/n-p-g催化剂上被更均匀地分散,其表现的活性比表面积最大。
4.3催化剂稳定性
电极催化剂的稳定性是燃料电池得以应用的关键因素之一。pd/n-p-g、pd/p-n-g、pd/p-g、pd/n-g和pd/g对甲醇的电催化氧化反应的稳定性测试结果如图5所示,从图中可以看出5种催化剂的催化电流密度均在最初阶段发生快速的衰减,这是因为甲醇在催化剂表面不断被氧化,生成的反应中间产物(co,cho等)在其表面聚集吸附,引起催化剂中毒,减少了有效催化活性位点,从而阻碍其进一步氧化甲醇,所以电流密度会不断降低。随着时间延长最终达到稳定状态。从衰减的过程来看,在1750s之前,pd/n-p-g的电流密度一直高于pd/n-p-g的相应值,1750s之后,两者趋于一致。而pd/n-p-g和pd/n-p-g的电流密度在3600s的测试期内,始终高于pd/p-g、pd/n-g和pd/g催化剂的相应值。在3600s时,pd/n-p-g和pd/n-p-g的电流密度均为11.9ma·cm-2,分别是pd/p-g(2.45ma·cm-2)、pd/n-g(1.22ma·cm-2)和pd/g(0.85ma·cm-2)的4.9倍、9.8倍和14倍。说明n/p掺杂会促进催化剂的稳定性能提高,而pd/n-p-g稳定性能最佳。
5催化剂表征
5.1xrd图谱分析
选取了部分催化剂进行了xrd表征,结果见图6。从图中可以看到pd/p-g和pd/n-p-g这两个样品在2θ=25.8°(d(002)=0.344nm)和25.3°(d(002)=0.352nm)处左右出现了宽化的石墨c的(002)衍射峰,一方面显示出多层石墨烯的结构特点;另一方面相比较于pd/p-g,pd/n-p-g有更大的(002)晶面间距,说明通过n、p双掺杂后石墨烯片层团聚变少,从而能够提供更大的可利用比表面积,有利于催化剂活性组分的负载和分散。同时这两个催化剂在40.0°,46.4°,68.0°,82.0°处出现的衍射峰分别为pd的(111)、(200)、(220)和(311)晶面特征衍射峰,表明pd粒子为面心立方晶体结构(jcpdsno.46-1043)。另外,从衍射峰的强度可以看出负载的pd金属颗粒具有良好的结晶性。根据pd(111)晶面所对应的衍射峰,可利用scherrer公式d=0.89λ/βcosθ计算pd的平均粒径大小(其中,λ为x射线的波长,β为衍射峰对应的半峰宽(以弧度表示),θ为晶面所对应的角度),得到pd/p-g、pd/n-p-g和pd/p-n-g催化剂中pd的平均粒径分别为13.6nm、7.6nm和9.7nm。
5.2tem图谱分析
图7为pd/n-p-g、pd/p-n-g、pd/p-g和pd/g的tem图像。四个样品都可以观察到石墨烯呈无序、透明、褶皱的片层状结构,还可以看到pd颗粒的大小及分布情况。pd/g催化剂上pd颗粒较为均匀分布且呈球形状,颗粒大小在5-20nm,不过存在较为严重的团聚现象。pd/p-g催化剂上pd颗粒的团聚现象稍有改善。对比而言pd/n-p-g、pd/p-n-g催化剂pd颗粒分布相对更为均匀,也更为细小,颗粒约为5-10nm。说明n和p的掺杂有利于pd金属颗粒在石墨烯表面的分散。
5.3xps图谱分析
xps是一种有效的表面分析手段,可定性分析样品的元素和分子结构等信息以及半定量分析元素的组成。pd/n-p-g和pd/n-p-g两个催化剂的xps结果见图8,具体的元素分析拟合结果见表1。其中图8(a)为两个样品的全谱图,可以发现两者在结合能为134.0,286.0,340.0和530.0ev都出现了信号峰,这分别对应于p2p,c1s,pd3d和o1s的特征峰,并且pd/n-p-g样品还在结合能为400.0ev出现n1s的信号。这些表明p或是n、p成功地掺杂进入碳材料的结构中。元素分析结果显示p的掺杂量从原子分数1.81%(pd/p-g)增加到3.12%(pd/n-p-g),说明石墨烯经过n掺杂后利于后续的p掺杂。
pd/n-p-g的n1s谱线拟合图见图8(b),通过分析可知样品中含有四种类型的n,分别为吡啶型n(n1,398.4ev),吡咯型n(n2,399.7ev),石墨型n(n3,400.8ev)和氧化型n(n4,402.8ev),其含量分别为28.38%,36.64%,24.08%和10.90%。
对pd/p-g、pd/n-p-g和pd/p-n-g催化剂的p2p的xps谱进行分峰拟合发现,pd/p-g(图8(c))的p2p可分成两个峰,对应于p-c键(133.19ev)和p-o键(133.98ev),两个物种的原子百分比为38.09:61.91。pd/n-p-g和pd/p-n-g(图8(d)和8(e))样品中p-o键(134.54ev)原子百分比降低到35.5%和53.5%,而p-c键(133.16ev)的原子百分比增加到64.5%和46.5%,这说明与单一p掺杂的石墨烯相比,将石墨烯进行n、p双掺杂后会显著提高p-c相的比例,一方面是因为n是富电子原子,并且n的电负性(3.04)比c(2.55)更大,在对石墨烯进行n掺杂后,会使相邻c原子赋予正电性。而p虽然也是富电子原子,但是p的电负性(2.19)比c还小,p可以充当电子给体,因此在n掺杂的基础上再进行p掺杂时,有利于电子的分配均匀。这种电子的分配有利于骨架c原子的大π键的共轭作用,可以发挥n、p两者的协同作用。
pd/n-p-g和pd/p-n-g催化剂的pd3dxps谱分峰结果见图8(f)和8(g)。pd3d的xps谱线通过分峰处理可以得到三对峰,分别对应于pd0、pdo和pdcl2。pd/n-p-g样品中pd0(335.53、340.75ev)、pdo(335.96、341.3ev)和pdcl2(337.84、343.33ev)的含量百分比分别为57.0%、17.4%和25.6%。而pd/p-n-g催化剂上三者的含量百分比分别为38.83%、40.33%和20.84%。
通过比较pd/g、pd/p-g、pd/n-p-g和pd/p-n-g催化剂的pd3dxps谱图,见图8(h)所示。pd/g的pd3d的两个最大的信号峰出现在结合能为335.9和340.95ev处,而pd/p-g、pd/n-p-g和pd/p-n-g催化剂均偏向结合能低的335.6和340.9ev处。另外结合表1可以发现,pd/g中金属pd的含量和pd2+的含量分别为6.42%和63.03%,而pd/p-g和pd/p-n-g样品中金属pd的含量分别提高到34.68%和38.83%,而pd/n-p-g催化剂的金属pd的含量高达57.0%。这说明通过对石墨烯载体进行掺杂后,会显著影响石墨烯的电子结构。具体表现为不仅会降低pd的3d结合能,而且会提高样品中金属pd的含量。另外我们可以看出,pd/n-p-g样品具有最高的金属pd含量和最低的pd2+含量,这表明n和p具有协同作用,从而增加样品中金属pd的含量,也即表明双掺杂的pd/n-p-g和pd/p-n-g样品比单掺杂的pd/p-g样品具有更多的活性位点。
表1.对pd/g,pd/p-g和pd/n-p-g催化剂的xps谱峰拟合结果的比较
结论
本发明通过合成pd/n-p-g、pd/p-n-g、pd/p-g、pd/n-g和pd/g这5种催化剂并对它们进行了电化学表征,结果发现催化剂的催化活性和稳定性顺序依次如下:pd/n-p-g>pd/p-n-g>pd/p-g>pd/n-g>pd/g。因而得出结论,石墨烯经过n和p掺杂后,其载钯催化剂的催化性能均大大提高。双元素掺杂比单元素掺杂石墨烯载pd催化剂的催化活性及稳定性更好。此外,掺杂顺序会对催化剂催化性能产生影响,很明显先n后p掺杂石墨烯载pd催化剂的催化性能优于先p后n掺杂石墨烯载pd催化剂。
以上结果表明,相对于pd/p-g、pd/g和pd/n-g这三种催化剂,pd/n-p-g和pd/n-p-g都具有更好的催化活性和电化学稳定性,而pd/n-p-g催化剂性能最为优越。这是由于:1、石墨烯经过n和p共掺杂后显著改变了石墨烯表面的电子结构,有利于骨架c的大π键的形成与稳定,有利于pd在其表面的铆定,阻碍了pd纳米颗粒的团聚,从而提高催化剂中金属pd的含量,增加活性位点;2、pd的3d结合能降低有利于甲醇催化氧化过程中生成的反应中间产物的脱除,从而暴露更多的活性位点,最终提高了催化剂的催化活性和稳定性。
以上所述的实施例只是本发明的一种较佳的方案,并非对本发明作任何形式上的限制,在不超出权利要求所记载的技术方案的前提下还有其它的变体及改型。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种纺织纤维/石墨烯/SrTiO<sub>3</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/Bi<sub>2</sub>O<sub>3</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/Bi<sub>2</sub>WO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiVO<sub>4</sub>/BiPO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>MoO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>WO<sub>6</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>MoO<sub>6</sub>/BiPO<sub>4</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/Bi<sub>2</sub>GeO<sub>5</sub>复合环境催化材料的制备方法
- 一种纺织纤维/石墨烯/BiOBr/BiOI复合环境催化材料的制备方法