碳催化剂、电池电极和电池的制作方法
本发明涉及碳催化剂、电池电极和电池。
背景技术:
当前,作为燃料电池的电极的催化剂,而使用铂催化剂。然而,有许多问题需要解决。例如,铂的储量是有限的。在聚合物电解质燃料电池(pefc)中,铂的使用增加了成本。因此,已经开发了不使用铂的替代技术。
具体而言,例如,在专利文献1中,描述了用于燃料电池的电极催化剂,其由通过碳化含有过渡金属的离子交换树脂而获得的碳化材料形成,并且其中各自具有平均粒径为10至20nm的壳状结构的许多碳颗粒以非聚集状态而组装。
引文清单
专利文献[ptl1]jp2007-207662a
发明概述
技术问题
然而,迄今为止,难以获得具有优异的催化活性和优异的耐久性的碳催化剂。
鉴于上述问题而进行了本发明,并且本发明的目的之一是提供各自具有优异的催化活性和优异的耐久性的碳催化剂、电池电极和电池。
问题的解决方案
为了解决上述问题,一方面,根据本发明一个实施方案的碳催化剂包含碳结构,其中所述碳结构通过使用2θ为24.0°±4.0°处的衍射峰f宽的bragg(布拉格)角而计算的微晶尺寸lc落入0.80nm以上且1.20nm以下的范围内,所述衍射峰f宽是通过将在由使用cukα射线的粉末x射线衍射获得的x射线衍射图中在衍射角2θ为26°附近的衍射峰进行分离获得的。并且显示出,在包括在25至1200℃温度下测量二氧化碳解吸量的程序升温解吸方法中,在650至1200℃的温度范围内的二氧化碳解吸量为97μmol/g以下。根据本发明的一个实施方案,提供了一种具有优异的催化活性和优异的耐久性的碳催化剂。
为了解决上述问题,另一方面,根据本发明一个实施方案的碳催化剂包含碳结构,其中所述碳结构通过使用2θ为24.0°±4.0°处的衍射峰f宽的布拉格角而计算的微晶尺寸lc落入0.80nm以上且1.20nm以下的范围内,所述衍射峰f宽是通过将在由使用cukα射线的粉末x射线衍射获得的x射线衍射图中在衍射角2θ为26°附近的衍射峰进行分离获得的。并且显示出,在包括在25至1200℃温度下测量一氧化碳解吸量和二氧化碳解吸量的程序升温解吸方法中,在650至1200℃的温度范围内的一氧化碳解吸量和二氧化碳解吸量的总和为647μmol/g以下。根据本发明的一个实施方案,提供了一种具有优异的催化活性和优异的耐久性的碳催化剂。
为了解决上述问题,还是另一方面,根据本发明一个实施方案的碳催化剂包含碳结构,其中所述碳结构通过使用2θ为24.0°±4.0°处的衍射峰f宽的布拉格角而计算的微晶尺寸lc落入0.80nm以上且1.20nm以下的范围内,所述衍射峰f宽是通过将在由使用cukα射线的粉末x射线衍射获得的x射线衍射图中在衍射角2θ为26°附近的衍射峰进行分离获得的。并且显示出,在包括在25至1200℃温度下测量一氧化碳解吸量的程序升温解吸方法中,在650至1200℃的温度范围内的一氧化碳解吸量为549μmol/g以下。根据本发明的一个实施方案,提供了一种具有优异的催化活性和优异的耐久性的碳催化剂。
此外,所述碳催化剂可以包括碳结构,其显示出在包括在25至1200℃温度下测量一氧化碳解吸量和二氧化碳解吸量的程序升温解吸方法中,在650至1200℃的温度范围内的一氧化碳解吸量和二氧化碳解吸量的总和为647μmol/g以下。此外,所述碳催化剂可以包括碳结构,其显示出在包括在25至1200℃温度下测量一氧化碳解吸量的程序升温解吸方法中,在650至1200℃的温度范围内的一氧化碳解吸量为549μmol/g以下。
另外,所述碳催化剂可以包括碳结构,其显示出在通过拉曼光谱法而获得的拉曼光谱中,在1580cm-1附近的g带和在1360cm-1附近的d带之间的最小强度与g带的强度之比为0.30或更大和0.49或更小。此外,所述碳催化剂可以包括碳结构,其显示出在通过拉曼光谱法而获得的拉曼光谱中,在1360cm-1附近的d带的半高宽为179cm-1以下。此外,所述碳催化剂可以包括碳结构,其显示出在通过拉曼光谱法而获得的拉曼光谱中,在1580cm-1附近的g带的半高宽为80cm-1以下。
此外,所述碳催化剂可以包括金属。此外,所述碳催化剂可以包括通过bet法而测量的比表面积为800m2/g或更大的碳结构。另外,所述碳催化剂可以包括碳结构,其氮原子浓度与碳原子浓度的比率为2.0%以上,该氮原子浓度和碳原子浓度通过x射线光电子能谱法而测定。此外,所述碳催化剂可以包括通过基于燃烧方法的元素分析而测量的氮原子含量为1.5重量%以上的碳结构。
此外,当通过使用旋转环盘电极装置而进行电位扫描而获得的氧还原伏安图中,当–10μa/cm2的还原电流流过时,所述碳催化剂的电压可以为0.810v(相对于nhe)或更高,其中所述旋转环盘电极装置包括含有碳催化剂的工作电极。此外,当通过使用旋转环盘电极装置而进行电位扫描而获得的氧还原伏安图中,在施加0.7v(相对于nhe)的电压时,所述碳催化剂的电流密度可以为–1.0ma/cm2以下,其中所述旋转环盘电极装置包括含有碳催化剂的工作电极。
此外,关于在第五个循环的伏安图中,由在1.0v至0.0v的电位扫描时间下的曲线和在0.0v至1.0v的电位扫描时间下的曲线而包围的区域的面积,该伏安图是通过使用旋转环盘电极装置在0.0v至1.0v(相对于nhe)内进行五个周期的电位扫描而获得,其中所述旋转环盘电极装置包括含有碳催化剂的工作电极,在耐久性试验的每次开始时间和结束时间中,通过使用旋转环盘电极装置而将施加1.2v(相对于rhe)电压的状态持续10分钟,所述碳催化剂可以表现出5.70×10-5a·s以下的面积增加量,该面积增加量是通过从耐久性开始时的面积减去耐久性试验结束时的面积而获得。
此外,关于在通过使用旋转环盘电极装置进行电位扫描而得到的氧还原伏安图中,在施加0.7v(相对于nhe)的电压时的电流密度(ma/cm2),其中所述旋转环盘电极装置包括含有碳催化剂的工作电极,在耐久性试验的每次开始时间和结束时间中,通过使用旋转环盘电极装置而将施加1.2v(相对于rhe)电压的状态持续10分钟,所述碳催化剂可以表现出88.0%以上的电流密度维持率,所述电流密度维持率通过将耐久性结束时的电流密度除以耐久性试验开始时的电流密度而获得的值乘以100而得到。
为了解决上述问题,根据本发明的一个实施方案的电池电极包括上述碳催化剂中的任一种。根据本发明的一个实施方案,提供了一种包括具有优异的催化活性和优异的耐久性的碳催化剂的电池电极。
为了解决上述问题,根据本发明的一个实施方案的电池包括上述电池电极。根据本发明的一个实施方案,提供了一种包括具有优异的催化活性和优异的耐久性的碳催化剂的电池。
本发明的有益效果
根据本发明,提供了各自具有优异的催化活性和优异的耐久性的碳催化剂、电池电极和电池。
附图说明
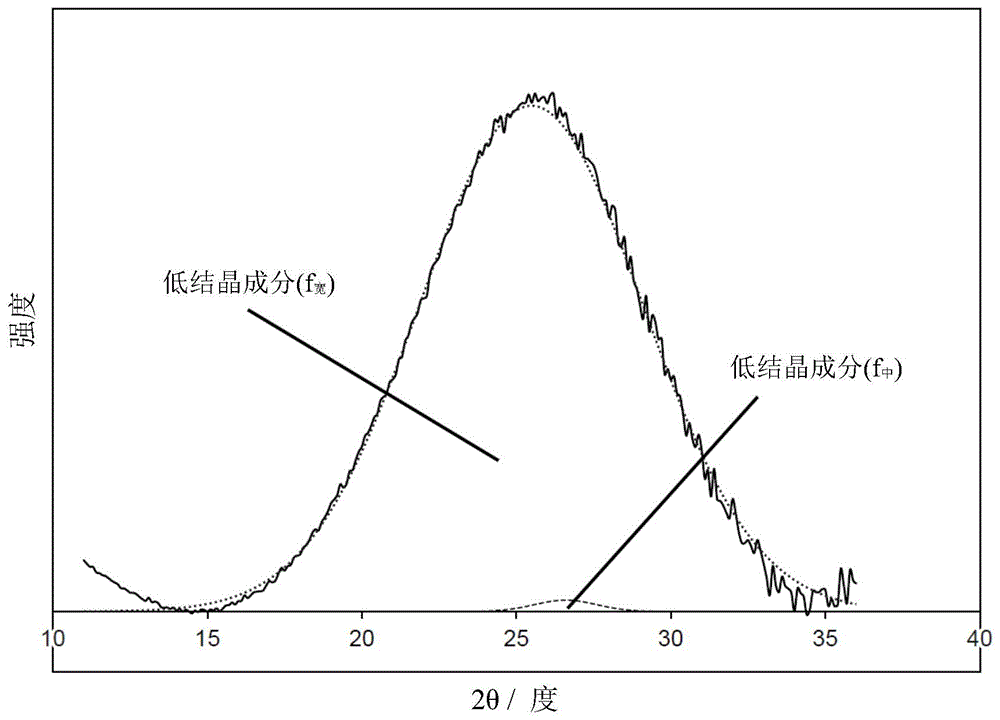
图1a是显示通过在根据本发明的一个实施方案的实施例1中获得的碳催化剂的粉末x射线衍射而得到的x射线衍射图中分离衍射峰而获得的结果的说明图。
图1b是显示通过在根据本发明的一个实施方案的实施例2中得到的碳催化剂的粉末x射线衍射而获得的x射线衍射图中分离衍射峰而得到的结果的说明图。
图1c是显示通过碳催化剂的粉末x射线衍射而获得的x射线衍射图中分离衍射峰而得到的结果的说明图。
图2是显示通过对本发明的一实施方案的实施例1中得到的碳催化剂的拉曼光谱而得到的拉曼光谱进行分析而得到的结果的说明图。
图3是显示通过对使用旋转环盘电极装置而得到的伏安图进行分析而得到的结果的说明图,其中所述旋转环盘电极装置包括工作电极,其中所述工作电极含有本发明的一个实施方案的实施例1中得到的碳催化剂。
图4a是显示通过使用旋转环盘电极装置而获得的氧还原伏安图的说明图,其中所述旋转环盘电极装置包括工作电极,其中所述工作电极含有本发明的一个实施方案的实施例1中得到的碳催化剂。
图4b是以放大的方式显示的由图4a的虚线而包围的部分x的说明图。
图5是显示通过对本发明的一个实施方案的实施例1至17和比较例1至4而得到的碳催化剂进行评估而获得的结果的说明图。
图6是显示通过对本发明的一个实施方案的实施例的空气电池的最大输出密度进行评估而得到的结果的说明图。
图7是显示通过对本发明的一个实施方式的实施例的空气电池的输出维持率进行评估而得到的结果的说明图。
具体实施方式
现在,将对根据本发明一个实施方案的碳催化剂(以下称作“本发明的催化剂”)、根据本发明一个实施方案的电池电极(以下称作“本发明的电极”),和根据本发明一个实施例的电池(以下称作“本发明的电池”)进行描述。本发明并不限于在该实施方案中所描述的实施例。
本发明的发明人对获得具有优异的催化活性和优异的耐久性的碳催化剂的技术手段进行了广泛的研究,结果,独特地发现了微晶尺寸lc具有特殊范围,并且在程序升温解吸方法中在特定温度范围内一氧化碳解吸量和/或二氧化碳解吸量等于或小于特定阈值的碳催化剂具有优异的催化活性和优异的耐久性,因此完成本发明,其中所述微晶尺寸lc通过粉末x射线衍射而获得。
即在一个方面,根据本发明一个实施方案的碳催化剂包含碳结构,其中所述碳结构通过使用2θ为24.0°±4.0°处的衍射峰f宽的布拉格角而计算的微晶尺寸lc落入0.80nm以上且1.20nm以下的范围内,所述衍射峰f宽是通过将在由使用cukα射线的粉末x射线衍射获得的x射线衍射图中在衍射角2θ为26°附近的衍射峰进行分离获得的,并且显示出,在包括在25至1200℃温度下测量co2解吸量的程序升温解吸方法(下文称作“tpd”)中,在650至1200℃的温度范围内的二氧化碳(co2)解吸量为97μmol/g以下。
当碳催化剂的碳结构具有由有助于碳催化剂的催化活性的弯曲碳网络平面而形成的层压结构时,在使用cukα射线的xrd图中,碳(002)衍射线出现在衍射角2θ为26°附近(例如在23.0°或更大且27.0°或更小的范围内)。该碳(002)衍射线可以包括衍生于作为高结晶组分的石墨结构的(002)衍射线和各自衍生于低结晶组分的一个或两个(002)衍射线。
具体而言,通过峰分离,可以将在衍射角(2θ)为26°附近的碳(002)衍射峰分离为三个衍射峰,即,低结晶组分的两个衍生峰f宽和f中以及高结晶组分的衍射峰f窄。
具体而言,宽峰f宽定义为衍射角2θ为24.0°±4.0°且半高宽为10.0°±7.0°的衍射峰。中峰f中定义为衍射角2θ为26.2°±0.3°且半高宽为2.0°±0.99°的衍射峰。窄峰f窄定义为衍射角2θ为26.5°±0.5°且半高宽为0.3°±0.2°的衍射峰。
通过峰分离并不是总会获得所有三个衍射峰。在这三个衍射峰中,仅可以获得两个衍射峰(例如,仅低结晶组分的两个衍射峰f宽和f中)。在三个衍射峰中,仅可以获得一个衍射峰(例如,仅低结晶组分的一个衍射峰f宽)。
对通过峰分离而获得的宽峰f宽进行分析以计算微晶尺寸lc。具体而言,通过将由峰分离而获得的宽峰f宽的布拉格角代入如下scherrer方程:lc=kλ/βcosθ而计算微晶尺寸lc。在scherrer方程式中,k代表scherrer常数(0.94),λ代表cukα射线的波长(0.15418nm),β代表半高宽(弧度),θ代表布拉格角(弧度)。
优选,本发明催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内。本发明催化剂的碳结构的微晶尺寸lc落在上述特定范围内的事实有助于本发明催化剂的优异催化活性。具体而言,当碳催化剂的碳结构中所含的活性点的数目增加时,碳结构的网络平面之间的相互作用则减弱。因此,网络平面的层数减少,结果碳结构的微晶尺寸lc则变小。因此,本发明催化剂的碳结构的微晶尺寸lc落在上述特定范围内,特别是等于或小于上述特定上限值的事实表明碳结构中所含的活性点的数目较大,即,本发明催化剂的催化活性高。
此外,本发明催化剂的碳结构在tpd中在650至1200℃的温度范围内co2解吸量优选90μmol/g以下,更优选86μmol/g以下,还更优选80μmol/g以下,特别优选70μmol/g以下。本发明催化剂的碳结构可以通过适当组合而限定:上述微晶尺寸lc的范围之一;和co2解吸量的上述阈值之一。
具体而言,例如优选,本发明催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内,并且co2的解吸量为90μmol/g以下,更优选86μmol/g以下,还更优选80μmol/g以下,特别优选70μmol/g以下。
当本发明催化剂的碳结构的微晶尺寸lc落入上述特定范围内且并且在tpd中在650至1200℃的温度范围内的co2解吸量等于或小于上述特定的阈值时,本发明的催化剂具有优异的催化活性和优异的耐久性。对于本发明催化剂的co2解吸量的下限值没有特别限制。本发明催化剂的碳结构在在tpd中在650至1200℃的温度范围内的co2解吸量为5μmol/g以上。
在另一个方面,根据本发明一个实施方案的碳催化剂包含碳结构,其中所述碳结构通过使用2θ为24.0°±4.0°处的衍射峰f宽的布拉格角而计算的微晶尺寸lc落入0.80nm以上且1.20nm以下的范围内,所述衍射峰f宽是通过将在由使用cukα射线的粉末xrd获得的xrd衍射图中在衍射角2θ为26°附近的衍射峰进行分离获得的,并且显示出,在包括在25至1200℃温度下测量co解吸量和co2解吸量的tpd中,在650至1200℃的温度范围内的一氧化碳(co)解吸量和二氧化碳(co2)解吸量的总和(下文称作“(co+co2)解吸量”)为647μmol/g以下。
还在该情况下,本发明的催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内。此外,本发明的催化剂的碳结构在tpd中在650至1200℃的温度范围内的(co+co2)解吸量优选635μmol/g以下,更优选620μmol/g以下,还更优选600μmol/g以下,特别优选580μmol/g以下。本发明催化剂的碳结构可以通过适当组合而限定:上述微晶尺寸lc的范围之一;和(co+co2)解吸量的上述阈值之一。
具体而言,例如优选本发明的催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内,并且(co+co2)解吸量为635μmol/g以下,更优选620μmol/g以下,还更优选600μmol/g以下,特别优选580μmol/g以下。
当本发明催化剂的碳结构的微晶尺寸lc落入上述特定范围内且并且在tpd中在650至1200℃的温度范围内的(co+co2)解吸量等于或小于上述特定的阈值时,本发明的催化剂具有优异的催化活性和优异的耐久性。对于本发明催化剂的(co+co2)解吸量的下限值没有特别限制。本发明催化剂的碳结构在在tpd中在650至1200℃的温度范围内的(co+co2)解吸量可以为45μmol/g以上。
此外,本发明的催化剂具有微晶尺寸lc为0.80nm以上且1.20nm以下的范围内的碳结构,并且显示出,在包括在25至1200℃温度下测量co解吸量和co2解吸量的程序升温解吸方法中,在650至1200℃的温度范围内的co2解吸量为97μmol/g以下,且在650至1200℃的温度范围内(co+co2)解吸量为647μmol/g以下。此时,本发明催化剂的碳结构可以通过适当组合而限定:上述微晶尺寸lc的范围之一;co2解吸量的上述阈值之一;和(co+co2)解吸量的上述阈值之一。
具体而言,例如优选本发明的催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内,和co2解吸量为90μmol/g以下,并且co解吸量与co2解吸量之和为635μmol/g以下,更优选co2解吸量为86μmol/g以下,并且co解吸量与co2解吸量之和为620μmol/g以下,还更优选co2解吸量为80μmol/g以下,并且co解吸量与co2解吸量之和为600μmol/g以下,特别优选co2解吸量为70μmol/g以下,并且co解吸量与co2解吸量之和为580μmol/g以下。
在另一个方面,根据本发明一个实施方案的碳催化剂包含碳结构,其中所述碳结构通过使用2θ为24.0°±4.0°处的衍射峰f宽的布拉格角而计算的微晶尺寸lc落入0.80nm以上且1.20nm以下的范围内,所述衍射峰f宽是通过将在由使用cukα射线的粉末xrd获得的xrd衍射图中在衍射角2θ为26°附近的衍射峰进行分离获得的,并且显示出,在包括在25至1200℃温度下测量co解吸量量的tpd中,在650至1200℃的温度范围内的co解吸量为549μmol/g以下。
还在该情况下,优选,本发明的催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内。此外,本发明的催化剂的碳结构在tpd中在650至1200℃的温度范围内的co解吸量优选547μmol/g以下,特别优选545μmol/g以下。本发明催化剂的碳结构可以通过适当组合而限定:上述微晶尺寸lc的范围之一;和co解吸量的上述阈值之一。
具体而言,例如优选本发明催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内,并且co解吸量为547μmol/g以下,特别优选545μmol/g以下。
当本发明催化剂的碳结构的微晶尺寸lc落入上述特定范围内且并且在tpd中在650至1200℃的温度范围内的co解吸量等于或小于上述特定的阈值时,本发明的催化剂具有优异的催化活性和优异的耐久性。对于本发明催化剂的co解吸量的下限值没有特别限制。本发明催化剂的碳结构在在tpd中在650至1200℃的温度范围内的co解吸量可以为40μmol/g以上。
此外,本发明的催化剂具有微晶尺寸lc为0.80nm以上且1.20nm以下的范围内的碳结构,并且显示出,在包括在25至1200℃温度下测量co解吸量和co2解吸量的tpd中,在650至1200℃的温度范围内的co解吸量为549μmol/g以下,且在650至1200℃的温度范围内co2解吸量为97μmol/g以下。此时,本发明催化剂的碳结构可以通过适当组合而限定:上述微晶尺寸lc的范围之一;co解吸量的上述阈值之一;和co2解吸量的上述阈值之一。
具体而言,例如优选本发明催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内,和co解吸量为547μmol/g以下,且co2解吸量为90μmol/g以下,更优选co解吸量为547μmol/g以下,且co2解吸量为86μmol/g以下,还更优选co解吸量为545μmol/g以下,且co2解吸量为80μmol/g以下,特别优选co解吸量为545μmol/g以下,且co2解吸量为70μmol/g以下。
此外,本发明的催化剂可以具有微晶尺寸lc为0.80nm以上且1.20nm以下的范围内的碳结构,并且显示出,在包括在25至1200℃温度下测量co解吸量和co2解吸量的程序升温解吸方法中,在650至1200℃的温度范围内的co解吸量为549μmol/g以下,且在650至1200℃的温度范围内(co+co2)解吸量为647μmol/g以下。此时,本发明催化剂的碳结构可以通过适当组合而限定:上述微晶尺寸lc的范围之一;co解吸量的上述阈值之一;和(co+co2)解吸量的上述阈值之一。
具体而言,例如优选本发明催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内,和co解吸量为547μmol/g以下,且(co+co2)解吸量为635μmol/g以下,更优选co解吸量为547μmol/g以下,且(co+co2)解吸量为620μmol/g以下,还更优选co解吸量为545μmol/g以下,且(co+co2)解吸量为600μmol/g以下,特别优选co解吸量为545μmol/g以下,且(co+co2)解吸量为580μmol/g以下。
此外,本发明的催化剂可以具有微晶尺寸lc为0.80nm以上且1.20nm以下的范围内的碳结构,并且显示出,在包括在25至1200℃温度下测量co解吸量和co2解吸量的程序升温解吸方法中,在650至1200℃的温度范围内的co解吸量为549μmol/g以下,且在650至1200℃的温度范围内co2解吸量为97μmol/g以下且在650至1200℃的温度范围内(co+co2)解吸量为647μmol/g以下。此时,本发明催化剂的碳结构可以通过适当组合而限定:上述微晶尺寸lc的范围之一;co解吸量的上述阈值之一;co2解吸量的上述阈值之一;和(co+co2)解吸量的上述阈值之一。
具体而言,例如优选本发明催化剂的碳结构的微晶尺寸lc为0.90nm以上且1.20nm以下的范围内,和co解吸量为547μmol/g以下,co2解吸量为90μmol/g以下,且(co+co2)解吸量为635μmol/g以下,更优选co解吸量为547μmol/g以下,co2解吸量为86μmol/g以下,且(co+co2)解吸量为620μmol/g以下,还更优选co解吸量为545μmol/g以下,co2解吸量为80μmol/g以下,且(co+co2)解吸量为600μmol/g以下,特别优选co解吸量为545μmol/g以下,co2解吸量为70μmol/g以下,且(co+co2)解吸量为580μmol/g以下。
在tpd中,将碳催化剂加热,并在25至1200℃的温度范围内测量来自碳催化剂的一氧化碳(co)的解吸量和/或二氧化碳(co2)的解吸量,不进行包括使官能团解吸的热处理和随后的氧吸附的预处理。
作为本发明催化剂的特征之一而限定的tpd中的co解吸量和/或co2解吸量反映了本发明催化剂的碳结构中所含的含氧官能团的质量和量。具体而言,本发明的催化剂的碳结构具有少量的含氧官能团,从而在上述tpd中的特定温度范围内的co解吸量和/或co2解吸量等于或小于上述特定阈值。本发明催化剂的碳结构中的含氧官能团的含量较少的事实有助于本发明催化剂的优异耐久性。
具体而言,认为所述碳结构的电化学氧化降解通过以含氧官能团的顺序氧化反应作为起点而进行。因此,认为可以用作氧化降解的起点的碳结构中含氧官能团的含量降低时,碳结构中降解起点的数量会减少,有助于改善碳结构的耐久性。在这方面,本发明的发明人已经发现,在含氧官能团中,特别是特定的含氧官能团会显著影响碳催化剂的耐久性,其中所述特定的含氧官能团会导致tpd中在650至1200℃的特定温度范围内的co解吸和/或co2的解吸。
本发明的催化剂可以具有碳结构,该碳结构在通过拉曼光谱法而获得的拉曼光谱中,在1,580cm-1附近的g带和在1,360cm-1附近的d带之间的最小强度与g带强度的比例(下文称作“iv/ig比例”)为0.30以上且0.49以下。
此时,在本发明催化剂的通过拉曼光谱法而获得的拉曼光谱中,在1,580cm-1附近(例如,1,550cm-1以上且1,650cm-1以下)检测到g带,并且在1,360cm-1(例如1,250cm-1以上且1,400cm-1以下)附近处检测到d带。g带和d带之间的最小强度iv与g带的强度ig的比例而计算的iv/ig比例为0.30以上且0.49以下。
此时,在拉曼光谱中,g带是源自理想石墨结构的组分,而d带是源自含有缺陷和边缘的弯曲结构的组分。g带和d带之间的最小强度iv取决于衍生于无定形结构的组分。因此,iv/ig比例是无定形结构的量与理想石墨结构的量的比例。在碳结构中,活性点存在于无定形结构中。当无定形结构的量太大时,碳催化剂易于降解。因此,认为iv/ig比例具有最佳范围。在这方面,本发明催化剂的碳结构的iv/ig比例落在上述特定范围内的事实有助于本发明催化剂的优异催化活性。
本发明催化剂可以具有在通过拉曼光谱而获得的拉曼光谱中在1,360cm-1附近的d带的半高宽为179cm-1以下的碳结构。此时,在本发明催化剂的通过拉曼光谱法而获得的拉曼光谱中,在1,360cm-1附近(例如1,250cm-1以上或1,400cm-1以下)检测到d带的半高宽为179cm-1以下。
通过下式而计算d带的半高宽:d带的半高宽(cm-1)=(ad–bd)×2(在该式中,ad表示对应于d带的强度id(d带的峰顶)的波数(cm-1),bd代表对应于低于ad的波数侧的d带强度id的一半的拉曼光谱的波数(cm-1))。
此时,在拉曼光谱中,d带的半高宽显示出碳结构中所含的弯曲结构的结晶性。具体而言,d带的半高宽较小意味着弯曲结构的结晶度高。因此,本发明催化剂的碳结构的d带半高宽等于或小于上述特定阈值的事实意味着所述碳结构含有高结晶度的弯曲结构。然后,本发明催化剂具有含有高结晶度的弯曲结构的碳结构的事实有助于本发明催化剂的优异耐久性。对本发明催化剂的d带半高宽的下限值没有特别限制,但d带的半高宽可以为80cm-1以上。
本发明的催化剂可以具有在通过拉曼光谱法而获得的拉曼光谱中的1580cm-1附近的g带的半高宽为80cm-1以下的碳结构。此时,在本发明的催化剂通过拉曼光谱法而获得的拉曼光谱中,在1580cm-1附近(例如1550cm-1以上且1,650cm-1以下)检测到g带的半高宽为80cm-1以下。
通过下式而计算g带的半高宽:g带的半高宽(cm-1)={(ag–bg)的绝对值}×2(在该式中,ag表示对应于g带的强度ig(g带的峰顶)的波数(cm-1),并且bg代表对应于高于ag的波数侧的g带强度ig的一半的拉曼光谱的波数(cm-1))。
此时,在拉曼光谱中,g带的半高宽显示出碳结构中所含的石墨结构的结晶性。具体而言,g带的半高宽较小意味着石墨结构的结晶度高。因此,本发明催化剂的碳结构的d带半高宽等于或小于上述特定阈值的事实意味着所述碳结构含有高结晶度的石墨结构。然后,本发明催化剂具有含有高结晶度的石墨结构的碳结构的事实有助于本发明催化剂的优异耐久性。对本发明催化剂的g带半高宽的下限值没有特别限制,但g带的半高宽可以为40cm-1以上。
本发明的催化剂可以包含金属。此时,对本发明催化剂中所含的金属没有特别的限制,只要能够获得本发明催化剂的上述特征,但是金属优选过渡金属。此外,本发明催化剂优选含有两种或更多种的金属,更优选含有两种或更多种的过渡金属。
在该实施方案中,过渡金属是属于元素周期表中的iii至xii族的金属,优选属于元素周期表中的iii至xii族的第四周期的过渡金属。具体而言,本发明催化剂中所含的过渡金属例如可以是选自钪(sc)、钛(ti)、钒(v)、铬(cr)、锰(mn)、铁(fe)、钴(co)、镍(ni)、铜(cu)、锌(zn)、钇(y)、锆(zr)、铌(nb)、钼(mo)、钌(ru)、铑(rh)、钯(pd)、银(ag)、镧系元素(例如,选自钕(nd)、钐(sm)和钆(gd)的一种或多种),和锕系元素的一种或几种,或者选自sc、ti、v、cr、mn、fe、co、ni、cu、zn、y、zr、nb、mo、ag、镧系元素(例如选自nd、sm和gd的一种或几种),和锕系元素,或者可以是选自所述组中的两种或更多种。
此外,本发明的催化剂优选含有选自ti、cr、fe、zn、nd、sm和gd的一种或多种,更优选选自所述组中的两种或更多种。此时,本发明的催化剂可以含有选自fe和zn中的一种或多种,或者可以含有fe和zn。
当本发明的催化剂含有上述特定的过渡金属时,本发明的催化剂还可以含有其他过渡金属。即,例如当本发明的催化剂含有选自ti、cr、fe、zn、nd、sm和gd中的一种或多种或者两种或更多种第一种过渡金属时,本发明催化剂还可以含有一种或多种选自sc、ti、v、cr、mn、fe、co、ni、cu、zn、y、zr、nb、mo、ru、rh、pd、ag、镧系元素(例如选自nd、sm和gd中的一种或多种)和锕系元素的第二种过渡金属,或者选自sc、ti、v、cr、mn、fe、co、ni、cu、zn、y、zr、nb、mo、ag、镧系元素(例如选自nd、sm和gd中的一种或多种)和锕系元素的第二种过渡金属,其不同于所述第一种过渡金属。
此外,本发明的催化剂可以不含铂(pt)。本发明的催化剂可以不含选自铂(pt)、钌(ru)、铑(rh)、钯(pd)、铱(ir)、金(au)和(os)的一种或多种。
当本发明的催化剂含有衍生于后面所述的用于碳化的原料的金属时,本发明的催化剂在其中含有金属,该金属已经包含在用于碳化的原料中。具体而言,即使通过后面所述的金属去除处理而生产本发明的催化剂的时候,本发明的催化剂中留有微量的源自原料的金属。
具体而言,例如本发明催化剂所含的金属为颗粒时,当形成本发明的催化剂的颗粒进行切割时,在通过切割而暴露的颗粒的截面上检测到金属。本发明催化剂中所含的金属可以例如通过感应耦合等离子体(icp)发射分光光度法而进行检测。
本发明的催化剂通过bet法而测量比表面积可以为800m2/g以上。此时,本发明的催化剂的比表面积优选1,000m2/g以上,特别优选1200m2/g以上。
本发明催化剂的比表面积等于或大于上述特定阈值的事实有助于提高通过本发明催化剂的化学反应的效率,并有助于优异的催化活性。对本发明催化剂的比表面积的上限值并没有特别限制,但是比表面积可以为3,000m2/g以下。
本发明的催化剂可以具有碳结构,所述碳结构显示出氮原子浓度(原子%)与碳原子浓度(原子%)的比例(下文称作“n/c比”)为1.5%以上。通过x射线光电子能谱法(下文称作“xps”)而测定氮原子浓度和碳原子浓度。此时,本发明催化剂的碳结构的n/c比优选1.8%以上,特别优选2.0%以上。
本发明催化剂的碳结构的n/c比等于或大于上述特定阈值的事实意味着该碳结构在其表面上含有大量的含氮官能团,这有助于本发明催化剂的优异催化活性。尽管对本发明催化剂的n/c比的上限值并没有特别限制,但是n/c比可以为15.0%以下。
本发明的催化剂可以具有碳结构,所述碳结构显示出基于燃烧方法的元素分析而测量的氮原子含量为1.5重量%以上。此时,本发明催化剂的碳结构的氮原子含量优选1.8重量%以上,特别优选2.0重量%以上。
本发明催化剂的碳结构的氮原子含量等于或大于上述特定阈值的事实意味着该碳结构含有大量的含氮官能团,这有助于本发明催化剂具有优异的催化活性。尽管对本发明催化剂的氮原子含量的上限值并没有特别限制,但是氮原子含量可以为15重量%以下。
本发明的催化剂可以具有1.0μm以下的平均粒径。本发明催化剂的平均粒径等于或小于上述特定阈值的事实有助于提高通过本发明催化剂的化学反应的效率,并有助于本发明催化剂的优异催化活性,还有助于提高包含本发明催化剂的电池电极的生产效率。对本发明催化剂的平均粒径的下限值并没有特别限制,所述平均粒径可以为0.050μm以上。
本发明催化剂可以具有1,000.0μm以下的最大粒径。此时,本发明催化剂的最大粒径例如优选为50.0μm以下,特别优选10.0μm以下。具体而言,例如,当本发明催化剂是用于燃料电池的碳催化剂(例如,用于燃料电池的阴极或阳极的碳催化剂,优选用于燃料电池的阴极的碳催化剂)时,本发明催化剂的最大粒径优选50.0μm以下,特别优选10.0μm以下。另外,在这些情况下,虽然对本发明催化剂的最小粒径并没有特别限制,但是最小粒径可以为0.001μm以上。
在通过使用具有包含本发明碳催化剂的工作电极的旋转环盘电极装置实施扫描施加电位而获得的氧还原伏安图中在流过–10μa/cm2的还原电流时,本发明催化剂的电压(下文称作“氧还原起始电位(eo2)”)为0.810v(相对于nhe)以上。
本发明的催化剂的氧还原起始电位eo2等于或大于上述特定阈值的事实支持了本发明催化剂具有优异的催化活性。尽管对本发明催化剂的氧还原起始电位eo2的上限值并没有特别限制,但是电位eo2可以为1.0v(相对于nhe)以下。
在通过使用具有包含本发明碳催化剂的工作电极的旋转环盘电极装置实施扫描施加电位而获得的氧还原伏安图中施加有0.7v(相对于nhe)的电压时,本发明催化剂的电流密度(i0.7)可以为–1.0ma/cm2以下。
本发明催化剂的电流密度i0.7(ma/cm2)等于或小于上述特定阈值的事实支持本发明催化剂具有优异的催化活性。尽管对本发明催化剂的电流密度i0.7(ma/cm2)的下限值并没有特别限制,但是电流密度i0.7可以为–5.0ma/cm2以上。
关于在第五个循环的伏安图中由从1.0v至0.0v扫描施加电位时的曲线和从0.0v至1.0v扫描施加电位时的曲线而包围的区域的面积(下文称作“cv面积”),本发明催化剂显示出面积增加量(下文称作“cv面积增加量”)为5.70×10–5a·s以下,其中所述伏安图是通过在耐久性测试的起始时和结束时使用具有包含本发明催化剂的工作电极的旋转环盘电极装置在从0.0v至1.0v(相对于nhe)的范围内进行五个循环的扫描施加电位而获得,其中在所述耐久性测试中通过使用所述旋转环盘电极装置保持施加1.2v(相对于rhe)电压的状态10分钟,所述面积增加量是通过由耐久性测试开始时的面积减去所述耐久性测试结束时的面积而获得。
此时,本发明催化剂的cv面积增加量优选5.50×10-5a·s以下,5.30×10-5a·s以下,特别优选5.10×10-5a·s以下。
此时,cv区域显示包含碳催化剂的工作电极中的双电层的电容,并反映了碳催化剂中所含的细孔的状态及其表面官能团的信息。cv面积增加量较大的事实表明该碳催化剂进行了降解,碳催化剂中所含的细孔及其表面官能团发生了变化,并且包含所述碳催化剂的工作电极中的双电层的电容增加。因此,本发明催化剂的cv面积增加量等于或小于上述特定阈值的事实支持本发明催化剂具有优异的耐久性。对本发明催化剂的cv面积增加量的下限值并没有特别限制,但cv面积增加量可以为4.50×10-5a·s以上。
关于在氧还原伏安图中在施加0.7v(相对于nhe)电压时的电流密度i0.7(ma/cm2),本发明碳催化剂显示出电流密度i0.7保持率为88.0%以上,所述氧还原伏安图是通过在耐久性测试的起始时和结束时使用具有包含本发明催化剂的工作电极的旋转环盘电极装置实施扫描施加电位而获得,在所述耐久性测试中通过使用所述旋转环盘电极装置保持施加1.2v(相对于rhe)电压的状态10分钟,所述电流密度保持率是通过将所述耐久性测试结束时的电流密度除以所述耐久性测试开始时的电流密度所获得的值乘以100而获得。
此时,本发明催化剂的电流密度i0.7保持率优选89.0%以上,更优选90.0%以上,特别优选91.0%以上。
本发明催化剂的电流密度i0.7保持率等于或大于上述特定阈值的事实支持本发明催化剂具有优异的耐久性。尽管对本发明催化剂的电流密度i0.7保持率的上限值并没有特别限制,但是电流密度i0.7保持率可以为95%以下。
本发明催化剂由本身具有催化活性的碳材料而形成。形成本发明催化剂的碳材料例如为如下所述的通过使包含有机物质的原料碳化而获得的碳化材料。此外,当本发明催化剂由通过使包含有机物质和金属的原料碳化而获得的碳化材料形成时,所述金属包含在本发明催化剂的碳结构中。此时,认为本发明催化剂的催化活性主要归因于碳结构本身所含的活性点而不是金属。上述事实得到以下事实的支持,即,甚至在包含衍生于用于碳化的原料的金属的本发明催化剂进行金属去除处理以降低金属的含量时,在金属去除处理之后,本发明催化剂的催化活性与金属去除处理之前对比基本没有降低,并且与本发明的催化剂不同,事实是在碳化之后,在碳化材料的表面上载有金属的碳材料并不具有优异的催化活性,其中所述碳化材料是通过将含有有机物质但不含有金属的原料进行碳化而获得。本发明催化剂的催化活性,例如为氧化活性和/或还原活性,更具体而言为氧还原活性和/或氢氧化活性。
由于本发明催化剂本身具有催化活性,因此本发明催化剂可以不含pt,或者本发明的催化剂可以不含稀有金属,诸如pt。然而,本发明并不限于此,并且本发明催化剂可以用作负载稀有金属的载体。此时,本发明催化剂所负载的稀有金属例如为选自由pt、ru、rh、pd、ir、au和os中的一种或多种。
即使当本发明催化剂用作稀有金属的载体时,也用作未载有稀有金属的载体的本发明催化剂(在载有稀有金属之前的本发明催化剂)是本身具有催化活性的碳材料,即碳催化剂。
只要获得具有上述特性的本发明催化剂,对本发明催化剂的生产方法并没有特别限制。在该实施方案中,描述了包括在压力下使包含有机物质的原料碳化的方法。
对原料中所含的有机物质并没有特别限制,只要该有机物质可以碳化即可。具体而言,作为有机物质,例如,使用高分子量有机化合物(例如,诸如热固性树脂和/或热塑性树脂的树脂)和/或低分子量有机化合物。另外,生物质可以用作有机物质。
作为有机物质,优选使用含氮的有机物质。对含氮的有机物质并没有特别限制,只要该含氮的有机物质是包含在其分子中含有氮原子的有机化合物的有机物质即可。当本发明催化剂是通过使包含含氮有机物质的原料碳化而获得的碳化材料时,本发明催化剂的碳结构含有氮原子。
具体而言,可以用作所述有机物质的是:例如选自聚丙烯腈、聚丙烯腈-聚丙烯酸酯共聚物、聚丙烯腈-聚丙烯酸甲酯共聚物、聚丙烯腈-聚甲基丙烯酸共聚物、聚丙烯腈-聚甲基丙烯酸-聚甲基烯丙基磺酸共聚物、聚丙烯腈-聚甲基丙烯酸甲酯共聚物、苯酚树脂、聚糠醇、呋喃、呋喃树脂、酚醛树脂、三聚氰胺、三聚氰胺树脂、环氧树脂、含氮螯合树脂(例如,选自多胺型、亚氨基二乙酸型、氨基磷酸型和氨基甲基膦酸酯型树脂的一种或多种)、聚酰胺酰亚胺树脂、吡咯、聚吡咯、聚乙烯基吡咯、3–甲基聚吡咯、丙烯腈、聚偏二氯乙烯、噻吩、恶唑、噻唑、吡唑、乙烯基吡啶、聚乙烯基吡啶、哒嗪、嘧啶、哌嗪、吡喃、吗啉、咪唑、1–甲基咪唑、2–甲基咪唑、喹喔啉、苯胺、聚苯胺、琥珀酸二酰肼、己二酸二酰肼、聚砜、聚氨基双马来酰亚胺、聚酰亚胺、聚乙烯醇、聚乙烯醇缩丁醛、苯并咪唑、聚苯并咪唑、聚酰胺、聚酯、聚乳酸、聚醚、聚醚醚酮、纤维素、羧甲基纤维素、木质素、甲壳质、甲壳胺、沥青、褐煤、丝绸、羊毛、聚氨基酸、核酸、dna、rna、肼、酰肼、尿素、希夫碱、聚咔唑、聚双马来酰亚胺、三嗪、聚丙烯酸、聚丙烯酸酯、聚甲基丙烯酸酯、聚甲基丙烯酸、聚氨酯、聚酰胺胺和聚碳二亚胺的一种或多种。
对所述原料中的有机物的含量并没有特别限制,只要获得本发明的催化剂即可,例如可以为5重量%以上且90重量%以下,优选10重量%以上且80重量%以下。
用于碳化的原料可以进一步包含金属。具体而言,此时包含有机物质和金属和金属的原料在压力下进行碳化。当本发明催化剂由通过使包含有机物质和金属的原料进行碳化而获得的碳化材料而形成时,本发明催化剂含有金属。
原料中所含的金属(即,本发明催化剂中所含的金属)优选过渡金属。另外,原料优选包含两种或更多种的金属,更优选两种或更多种的过渡金属。
在该实施方案中,过渡金属是属于元素周期表中iii至xii族的金属,优选是属于元素周期表中的iii至xii族的第四周期的过渡金属。具体而言,原料中所含的过渡金属例如可以是选自sc、ti、v、cr、mn、fe、co、ni、cu、zn、y、zr、nb、mo、ru、rh、pd、ag、镧系元素(例如,选自nd、sm和gd中的一种或多种)和锕系元素,或sc、ti、v、cr、mn、fe、co、ni、cu、zn、y、zr、nb、mo、ag、镧系元素(例如,选自nd、sm和gd中的一种或多种)和锕系元素中的一种或多种,或者可以是选自其中的两种或更多种。
另外,原料优选包含选自ti、cr、fe、zn、nd、sm和gd中的一种或多种,更优选选自两种或多种。此时,原料可以包含选自fe和zn中的一种或多种,或者可以包含fe和zn。
当原料包含上述特定的过渡金属时,原料可以还含有其它过渡金属。也就是说,例如,当原料包含选自ti、cr、fe、zn、nd、sm和gd中的一种或多种或者两种或更多种第一过渡金属时,该原料还可以包含一种或多种选自sc、ti、v、cr、mn、fe、co、ni、cu、zn、y、zr、nb、mo、ru、rh、pd、ag、镧系元素(例如,选自nd、sm和gd中的一种或多种)和锕系元素,或者sc、ti、v、cr、mn、fe、co、ni、cu、zn、y、zr、nb、mo、ag、镧系元素(例如,选自nd、sm和gd中的一种或多种)和锕系元素的不同于所述第一过渡金属的第二过渡金属。
另外,所述原料可以不含pt。所述原料可以不含选自pt、ru、rh、pd、ir、au和os中的一种或多种。
作为原料中所含的金属,使用金属的简单物质和/或金属的化合物。作为金属化合物,可以使用选自例如金属盐、金属氧化物、金属氢氧化物、金属氮化物、金属硫化物、金属碳化物和金属络合物中的一种或多种。
尽管对原料中的金属含量(当使用两种或更多种金属时为两种或更多种金属的总含量)并没有特别限制,只要其含量在获得本发明的催化剂的范围内即可,所述含量例如可以为1重量%以上且90重量%以下,优选2重量%以上且80重量%以下。
通过在压力下加热原料并使原料保持在使原料碳化的温度(以下称作“碳化温度”)而进行碳化。对所述碳化温度并没有特别限制,只要原料进行碳化即可。碳化温度例如300℃以上。具体而言,此时,将包含有机物物的原料在300℃以上的温度下在压力下进行碳化。
另外,可以将碳化温度例如设定为700℃以上,优选900℃以上,更优选1000℃以上,特别优选1100℃以上。对碳化温度的上限值并没有特别限制。碳化温度例如为3,000℃以下。
升至碳化温度的升温速度例如为0.5℃/min以上且300℃/min以下。使原料保持在碳化温度下的时间例如为1秒以上且24小时以下,优选5分钟以上且24小时以下。所述碳化优选在惰性气体气氛,诸如氮气气氛中进行。具体而言,例如,优选在诸如氮气的惰性气体流下而进行碳化。
对在其中进行碳化的气氛的压力并没有特别限制,只要该压力高于大气压即可。所述压力例如是表压为0.05mpa以上的压力。另外,可以将以表压计而进行碳化的气氛的压力设定为0.15mpa以上,优选0.20mpa以上,更优选0.40mpa以上,特别优选0.50mpa以上。具体而言,此时,含有有机物质的原料在等于或大于上述阈值(mpa)的以表压计的压力下进行碳化。
本发明催化剂的生产方法可以还包括对通过上述碳化而获得的碳化材料进行进一步处理。具体而言,例如,可以对碳化的材料进行选自氨处理、氢处理和o-烷基化处理中的一种或多种处理。此时,将包含有机物的原料在压力下进行碳化,然后对通过碳化而获得的碳化材料进行选自氨处理、氢处理和o-烷基化中的一种或多种处理。
对所述氨处理并没有特别限制,只要氨处理包括使碳化材料与氨接触即可。具体而言,所述氨处理例如包括在含氨的气体气氛中将碳化材料进行热键。
尽管对含氨气体的氨含量并没有特别限制,只要氨含量落入获得氨处理的效果的范围内即可,所述氨含量例如可以为0.1体积%以上、1.0体积%以上或3.0体积%以上。
尽管对在氨处理期间用于加热碳化材料的温度并没有特别限制,只要该温度落入获得氨处理的效果的范围内即可,该温度例如可以是300℃以上,优选500℃以上,特别优选700℃以上。对加热温度的上限值并没有特别限定,例如可以为1300℃以下,优选1000℃以下。在氨处理过程中的加热温度的范围通过适当地组合而限定:上述下限值之一;和上述上限值之一。
对氢处理并没有特别限制,只要该氢处理包括使碳化材料与氢接触即可。具体而言,所述氢处理例如包括在含氢的气体气氛中加热碳化材料的处理。对含氢气体的氢含量并没有特别限制,只要氢含量落入获得氢处理效果的范围内即可,例如,所述氢含量可以为0.1体积%以上、1.0体积%以上或3.0体积%以上。
尽管对在氢处理期间用于加热碳化材料的温度并没有特别限制,只要该温度落入获得氢处理的效果的范围内即可,该温度例如可以是300℃以上,优选500℃以上,特别优选700℃以上。对加热温度的上限值并没有特别限定,例如可以为1300℃以上,优选1000℃以上。氢处理过程中的加热温度的范围通过适当组合而限定:上述下限值之一;上述上限值之一。
对o-烷基化处理并没有特别限制,只要该o-烷基化处理包括使碳化材料的碳结构中引起o-烷基化的处理即可。所述o-烷基化处理例如o-甲基化处理或o-乙基化处理。所述o-甲基化处理例如包括在惰性气体例如氮气的气氛下,在遮光条件下使碳化的材料与三甲基甲硅烷基重氮甲烷进行接触的处理。
另外,碳化的材料可以进行金属去除处理。此时,例如,将包含有机物质的原料在压力下进行碳化,然后将通过碳化而获得的碳化材料进行金属去除处理。另外,例如,将包含有机物的原料在压力下进行碳化,然后将通过碳化而获得的碳化材料进行金属去除处理,然后,对金属去除处理之后的碳化材料进行一种或多种选自氨处理、氢处理和o-烷基化处理中的处理。所述金属去除处理是包括减少源自碳化材料中所含的原材料的金属的量的处理。所述金属去除处理例如使用酸的洗涤处理和/或电解处理。
本发明的电极包括本发明的上述催化剂。具体而言,本发明的电极例如是负载本发明催化剂的电池电极。具体而言,本发明的电极例如是电池电极,其包括电极基材和负载在所述电极基材上的本发明催化剂。
这里,如上所述,本发明催化剂本身具有催化活性,因此本发明的电极可以不含pt或上述稀有金属,例如pt。然而,本发明并不限于此。本发明的电极例如可以包括,用作负载稀有金属的载体的本发明催化剂,以及由本发明催化剂负载的稀有金属。
本发明电极例如是燃料电池(例如聚合物电解质燃料电池)、空气电池、水电解电池(例如聚合物电解质水电解电池)、氧化还原液流电池或卤素电池的电极。另外,本发明电极例如是阴极或阳极,优选阴极。具体而言,本发明电极是燃料电池、空气电池、水电解电池、氧化还原液流电池或卤素电池的阴极或阳极,优选燃料电池阴极、空气电池阴极、水电解池阴极、氧化还原液流电池的阴极或卤素电池的阴极。
本发明的电池包括上述电池电极。具体而言,本发明的电池例如是包括本发明电极的燃料电池(例如聚合物电解质燃料电池)、空气电池、氧化还原液流电池或卤素电池。本发明的电池可以包括包含本发明电极的膜/电极组件。本发明的电池是包括本发明电极作为阴极或阳极的电池,优选包括本发明电极作为阴极的电池。具体而言,本发明电池是包括本发明电极作为阴极或阳极的燃料电池、空气电池、氧化还原液流电池或卤素电池,优选包含本发明电极作为阴极的燃料电池、空气电池、氧化还原液流电池或卤素电池。
下面,将描述根据该实施方案的具体实施例。
实施例
实施例1
将1.0克聚丙烯腈(pan)、1.0克2–甲基咪唑、6.0克氯化锌(zncl2)和30克二甲基甲酰胺进行混合。通过干燥而从获得的混合物中除去溶剂。将干燥的混合物在大气中加热以在250℃下变得不溶。
将25克的不溶混合物、0.03克的六水合氯化铁(iii)(fecl3·6h2o)和105克的二甲基甲酰胺进行混合。通过干燥而从获得的混合物中除去溶剂。
将干燥的混合物加热并在氮气气氛中在0.9mpa的表压下保持在1100℃以进行碳化。
将稀盐酸加入到通过碳化而获得的碳化材料中,然后搅拌。然后,使用膜过滤器过滤含有碳化材料的悬浮液,并用蒸馏水洗涤碳化材料直至滤液变为中性。因此,通过使用酸的洗涤进行了金属去除处理。
将金属去除处理后的碳化材料用粉碎机进行粉碎直至其平均粒径为1μm以下。因此,获得了粉碎的碳化材料作为实施例1的碳催化剂。
实施例2
将实施例1中获得的碳催化剂加热,并在使100%氨气以0.3l/min循环的气氛中在900℃下保持1小时。然后,氨气用氮气进行置换,并将碳催化剂在氮气气氛中在500℃下保持10分钟。获了得通过在氮气气氛中自然冷却而冷却的碳催化剂作为实施例2的碳催化剂。
实施例3
在氮气循环下将碳酸氢铵加热到150℃,从而产生包含氨、二氧化碳和水蒸气的混合气体。
将实施例1中获得的碳催化剂在氮气气氛中加热并在900℃下保持1小时,其中所述氮气气氛中,如上所述产生的混合气体在其中循环。然后,将混合气体用氮气进行置换,得到了通过在氮气氛下自然冷却而冷却的碳催化剂作为实施例3的碳催化剂。
实施例4
将实施例1中获得的碳催化剂加热并在100%氢气气氛中在900℃下保持1小时。然后,获得了通过在氢气气氛中自然冷却而冷却的碳催化剂作为实施例4的碳催化剂。
实施例5
将1.0克实施例1中获得的碳催化剂和100ml市售的超脱水甲醇进行混合,并且将得到的混合液在搅拌下用超声波处理2分钟。将超声处理后的混合液在遮光条件下在氮气气氛中与1.0m三甲基甲硅烷基重氮甲烷/己烷溶液混合并搅拌15小时。然后,通过干燥从混合液中除去溶剂。因此,获得了干燥后的碳催化剂作为实施例5的碳催化剂。
实施例6
以与实施例2相同的方式获得了实施例6的碳催化剂,不同之处在于,在不溶化之前而制备了还包含0.018克氯化铬六水合物(crcl3·6h2o)的混合物,并对混合物进行了不溶化。
实施例7
以与实施例3相同的方式而获得实施例7的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.018克氯化铬六水合物(crcl3·6h2o)的混合物,并对混合物进行不溶化。
实施例8
以与实施例2相同的方式而获得实施例8的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.06克的硼酸(b(ho)3)的混合物,并且对混合物进行不溶化。
实施例9
以与实施例2相同的方式而获得实施例9的碳催化剂,不同之处在于,使用2.0克的2–甲基咪唑代替1.0克的2–甲基咪唑。
实施例10
以与实施例2相同的方式获得实施例10的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.69克的氯化锗(iv)(gecl4)的混合物,并且对混合物进行不溶化。
实施例11
以与实施例2相同的方式获得实施例11的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.06克六水合氯化钐的混合物(smcl3·6h2o),并且对混合物进行了不溶化。
实施例12
以与实施例2相同的方式而获得实施例12的碳催化剂,不同之处在于,在不溶化之前制备了还含有0.06克六水合氯化钕的混合物(ndcl2·6h2o),并且对该混合物进行不溶化。
实施例13
以与实施例2相同的方式获得实施例13的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.06克六水合硝酸铅的混合物(pb(no3)2·6h2o),并且对该混合物进行了不溶化。
实施例14
以与实施例2相同的方式获得实施例14的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.075克六水合硝酸钆(gd(no3)3·6h2o)的混合物,并对混合物进行不溶化。
实施例15
以与实施例2相同的方式而获得实施例15的碳催化剂,不同之处在于,在不溶化之前制备了还包含1.06克的氯化铋(iii)(bicl3)的混合物,并对混合物进行了不溶化。
实施例16
以与实施例2相同的方式而获得实施例16的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.03克的氮化钛(tin)的混合物,并且对该混合物进行了不溶化。
实施例17
以与实施例2相同的方式获得实施例17的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.48克的氯化银(agcl)的混合物,并且对该混合物进行了不溶化。
比较例1
将1.0克聚丙烯腈(pan)、1.0克2–甲基咪唑、6.0克氯化锌(zncl2)和30克二甲基甲酰胺进行混合。通过干燥而从获得的混合物中除去溶剂。将干燥的混合物在大气中加热以在250℃下变得不溶。
将25克的不溶的混合物、0.03克的六水合氯化铁(iii)(fecl3·6h2o)和105克的二甲基甲酰胺进行混合。通过干燥而从获得的混合物中除去溶剂。
将干燥的混合物加热并在氮气气氛中在常压下在1,100℃保持1小时以使其碳化。将稀盐酸加入到通过碳化而获得的碳化材料中,然后搅拌。然后,使用膜过滤器对含有碳化材料的悬浮液进行过滤,并用蒸馏水洗涤碳化材料直至滤液变为中性。因此,通过使用酸的洗涤而进行了金属去除处理。
将金属去除处理后的碳化材料用粉碎机粉碎至其平均粒径为1μm以下。因此,获得了粉碎的碳化材料作为比较例1的碳催化剂。
比较例2
以与比较例1相同的方式而获得比较例2的碳催化剂,不同之处在于碳化是在1200℃下而不是1100℃下进行。
比较例3
以与比较例1相同的方式获得比较例3的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.018克氯化铬六水合物(crcl3·6h2o)的混合物,并对该混合物进行了不溶化。
比较例4
以与比较例2相同的方式获得比较例4的碳催化剂,不同之处在于,在不溶化之前制备了还包含0.018克氯化铬六水合物(crcl3·6h2o)的混合物,并且对该混合物进行了不溶化。
然后,对如上所述而获得的每种碳催化剂进行如下所述的分析。在以下每个分析的描述中而描述的所用碳催化剂的重量是指已经在真空中在80℃下热处理3小时的碳催化剂的重量。
粉末x射线衍射
进行了碳催化剂的粉末xrd测量。当在以通过粉末xrd获得的衍射图中分析碳(002)衍射线时,根据“117日本科学促进会委员会”的基于“通过x射线衍射法而测定碳材料的晶格参数和微晶尺寸的程序”进行校正。校正的细节描述于文献中(carbon,no.221,52-60页(2006))。
在“通过x射线衍射测量碳材料的晶格参数和微晶尺寸的程序”中,在每个预先计算的测量角度上,通过将每个测量角度的测量强度除以校正系数fct=l·p·a·fc2进行校正。此时,建立了l=1/(sin2θ·cosθ),p=(1+cos22θ·cos22θ′)/(1+cos22θ′),
以及a=[1–sin2θ/2μ′br][1–exp(–2μ′t/sinθ)]+(2t·cosθ/br)exp(–2μ′t/sinθ)
在上述表达式中,当使用反向单色仪时,θ表示测角计的角度,并且θ′表示单色仪晶体的衍射角(0°)。μ′表示样品的表观线性吸收系数(0.4219mm-1),并且br表示样品表面上的x射线的辐射宽度,并且由以下表达式给出:br=rsinβ。此时,β表示发散狭缝宽度(2/3°),r表示测角仪半径(285mm)。
另外,fc表示原子散射因子,并且通过以下表达式而得到:fc=(2.26069·exp(–0.226907·s2)+1.56165·exp(–0.00656665·s2)+1.05075·exp(–0.0975618·s2)+0.839259·exp(–0.555949·s2)+0.286977)。此时,建立了s=(sinθ)/λ。
在所获得的xrd图案中,对2θ为26°的衍射角附近的衍射峰进行峰分离。通过将重叠的衍射峰近似叠加到高斯基本波形上而进行峰分离。通过优化峰强度、半高宽以及用作各组分的高斯函数的峰位置作为参数,对经过背景校正的衍射图案进行拟合。通过将10°至20°的衍射角(2θ)附近与30°至40°的衍射角(2θ)附近连接的直线定义为背景并且从各个衍射强度中减去背景而进行背景校正。
通过将在2θ为26°衍射角附近的衍射峰(在2θ为26°衍射角附近具有峰顶的衍射峰)分离为如下三个衍射峰中的至少一个衍射峰而进行峰分离:f宽、f中、和f窄。
更具体而言,通过以下步骤而进行峰分离。在使用经过背景校正的cukα射线的x射线衍射图中,将具有在衍射角2θ为26°附近的峰顶的衍射峰近似地叠加到高斯基本波形上,因而优化了通过优化峰强度、半高宽和峰位置,上述衍射峰中所含的三个衍射峰中的至少一个(例如,当上述衍射峰中包含两个以上的衍射峰时,对两个或更多个重叠的衍射峰中的每一个)进行拟合,从而进行峰分离。
曲线拟合的进行,应使残差平方和变得最小。所述残差平方是指在每个测量的衍射角处的残差误差的平方,并且残差平方和是指残差平方的和。此外,残差误差是指在使用cukα射线的校正后的x射线衍射图案中,在衍射角2θ为26°附近具有峰顶的衍射峰的强度与通过分离而得到述三个衍射峰(f宽、f中、和f窄)中至少一个衍生峰的强度总和之差。通过该峰分离,获得上述三个衍射峰f宽、f中、和f窄中的至少一个衍射峰。
作为上述峰分离的实例,在图1a中示出了实施例1中获得的碳催化剂的xrd图,在图1b中示出了实施例2中获得的碳催化剂的xrd图。图1a和图1b中,横轴表示衍射角2θ(°),纵轴表示强度。
如图1a所示,通过在衍射角2θ为26°附近的衍射峰的峰分离,在实施例1中获得的碳催化剂中得到了低结晶组分的两个衍射峰f宽和f中。此外,如图1b所示,通过衍射角2θ为26°附近的衍射峰的峰分离,在实施例2中获得的碳催化剂中得到组分了低结晶组分的一个衍射峰f宽。
在上述峰分离中,如图1c所示,通过衍射角2θ为26°附近的衍射峰的峰分离,可以得到低结晶组分分的两个衍射峰f宽和f中和高结晶组分的一个衍射峰f窄。
然后,对通过上述峰分离获得的宽峰f宽进行分析,以计算微晶尺寸lc。具体而言,通过将由峰分离获得的宽峰f宽的布拉格角代入以下scherrer方程来计算微晶尺寸lc:lc=kλ/βcosθ。在所述scherrer方程中,k表示scherrer常数(0.94),λ表示cukα射线的波长(0.15418nm),β表示宽峰f宽的半高宽(弧度),θ表示f宽的布拉格角(弧度)。
程序升温解吸方法
通过tpd而测量碳催化剂的co解吸量和co2解吸量。具体而言,将碳催化剂安装在程序升温解吸装置(由microtracbelcorp.制造)中,并在高真空下进行热处理以解吸碳催化剂的表面官能团。然后,将氧吸附到碳催化剂的表面,并且使载气(he)以20ml/min而循环以加热碳催化剂。用四极质谱仪(qms)测量所解吸的气体。
具体而言,首先,将0.05克的碳催化剂填充到由石英制成的反应管的中心部分中,并在程序升温解吸装置中进行设置。使氦气(he)在装置中循环,并将装置内部在25℃下保持120分钟,从而使装置稳定。然后,将碳催化剂加热,以使温度以10℃/min的升温速率从25℃升至1200℃。在温度升高期间,氦气(he)以20ml/min的速度进行循环,并检测到解吸的气体。记录温度(横轴)和检测强度(纵轴)之间的关系。
然后,对650℃至1200℃的温度范围内解吸的气体的量进行测定。具体而言,计算650℃至1200℃的温度范围内的气体的检测强度(检测强度区域)的积分值。
同时,通过使用一氧化碳和氦的混合气体(一氧化碳:10体积%),二氧化碳和氦的混合气体(二氧化碳:10体积%)和氮气(氮气:100体积%)作为参考气体,获得了一氧化碳(质量数:28)、二氧化碳(质量数:44)和氮(质量数:14)的释放量之间的相关性的校准曲线,以及检测强度区域。
然后,基于通过测量而获得的检测强度面积和校准曲线,对从碳催化剂中的一氧化碳和二氧化碳的解吸量(释放量)进行测定。具体而言,通过将所得的一氧化碳和二氧化碳的释放量除以用于测量的碳催化剂的重量,而计算从碳催化剂中的一氧化碳和二氧化碳的解吸量(每碳催化剂的重量释放量)。关于一氧化碳,通过从质量数28的释放量减去质量数14(氮)的释放量而测定仅一氧化碳的释放量。
拉曼光谱
通过拉曼光谱而分析碳催化剂。通过使用horiba显微激光拉曼光谱仪(labram,horibajobinyvon)测量拉曼光谱。用于测量的激光器的激发波长为532nm且输出为50mw。在90秒的曝光以及通过中性密度滤光片d3的多次扫描的条件下而进行测量,以获得拉曼光谱。
在获得的拉曼光谱中,识别出在1,580cm-1附近的g带和在1,360cm-1附近的d带。此外,基于g带的强度ig(g带峰顶的峰强度)、d带的强度id(d带峰顶的强度)以及g带和d带之间的最小强度iv而获得g带的半高宽(cm-1)、d带的半高宽(cm-1)和iv/ig比。
作为上述拉曼光谱的一个实例,通过分析由实施例1中获得的碳催化剂的拉曼光谱获得的拉曼光谱而得到的结果示于图2中。在图2中,横轴表示波数(cm-1)。纵轴表示强度。虚线表示背景。ad代表与d带的峰顶相对应的波数(cm-1)。bd表示与拉曼光谱相对应的波数(cm-1),其中该拉曼光谱在比ad低的波数侧表现出d带强度id的一半的强度。ag表示与g带的峰顶相对应的波数(cm-1)。bg表示与拉曼光谱相对应的波数(cm-1),其中该拉曼光谱在比ag高的波数侧表现出g带强度ig的一半的强度。
比表面积
碳催化剂的比表面积通过使用比表面积/细孔分布测定装置(shimadzucorporation生产的tristar3000),使用氮气的bet法而测定。具体而言,首先,将0.1克的碳催化剂在100℃和6.7×10-2pa下保持3小时,从而去除吸附到碳催化剂上的水分。然后,通过bet法从77k下的氮吸附等温线而得到碳催化剂的比表面积(m2/g)。通过在77k的温度下测量与氮气压力的变化相关的对碳催化剂的氮吸附量的变化而得到77k下的氮吸附等温线。
x射线光电子能谱
通过xps而分析碳催化剂。具体而言,通过使用x射线光电子能谱仪(axisnova,ratos制造)测量来自碳催化剂表面上的碳原子和氮原子的每个核的光电子能谱。使用alkα线(10ma,15kv,通过能量:40ev)作为x射线源。在所获得的光电子光谱中,校正了结合能,使得源自碳原子的1s轨道的c1s峰的峰顶位于284.5ev。
基于每个光谱的峰面积和检测灵敏度系数而获得碳催化剂表面上的氮原子和碳原子的元素浓度(原子%)。然后,将氮原子浓度(原子%)除以碳原子浓度(原子%)而得到的值乘以100,从而计算出n/c比(%)。
元素分析
对该碳催化剂进行了元素分析。具体而言,通过使用有机微量元素分析装置(2400ii,perkinelmerco.,ltd.制造)通过燃烧法而测量碳催化剂的氮含量。在燃烧管温度为980℃,还原管温度为640℃的条件下,使用氦气作为载气对2mg的碳催化剂进行分析。然后,将氮的重量除以碳催化剂的总重量而得到的值乘以100,从而计算出氮原子含量(重量%)。
平均粒径
测量了碳催化剂的平均粒径。具体而言,使用纳米颗粒直径分布测定装置(shimadzucorporation,sald-7100h),通过激光衍射法而测定碳催化剂的粒径。更具体而言,首先,将一滴表面活性剂添加到10mg的碳催化剂中,并且将40克的蒸馏水添加到该混合物中,从而制备悬浮液。然后,将悬浮液进行均化器处理20分钟以制备分散体。将制备的分散液滴入流动池中,在流动池中使蒸馏水循环直至衍射/散射光强度的最大值变为50±5,从而测量粒径。由所得粒径分布(体积分布)测得的中位直径(d50)作为平均粒径得。在各自具有频率(%)为0.001以上的所得粒径分布(体积分布)中的粒径中,将最大值定义为最大粒径,将最小值定义为最小粒径。
催化剂活性和耐久性
通过使用旋转环盘电极装置(basinc.制造的rrde-3a旋转环盘电极装置ver.1.2)和双电化学分析仪(chi700c,由alscorporation制造)对碳催化剂的催化活性和耐久性进行了评估。具体而言,首先,制造具有包含碳催化剂的工作电极的三极旋转环盘电极装置。具体而言,将5mg碳催化剂、50μl的5%的nafion(商标)(sigma-aldrich制造的nafion,全氟离子交换树脂,5%溶液(产品编号:510211))、400μl的水和100μl的异丙醇进行混合以制备浆料。然后,对浆料进行超声处理10分钟,然后进行均化器处理2分钟。将得到的浆料施加于工作电极(rrde-3a的环状圆盘电极,铂环状金圆盘电极,圆盘直径4mm,bas公司制造)上,以使碳催化剂的施用量为0.1mg/cm2,然后干燥,因而制备了含有碳催化剂的工作电极。
此外,将铂电极(basinc.制造的23cm的pt对电极)用作对电极,并且将可逆氢电极(rhe)(由ecfrontierco.制造的存储型可逆氢电极)用作参比电极。因此,获得了具有含碳催化剂的工作电极、用作对电极的铂电极和用作参比电极的可逆氢电极(rhe)的旋转环形盘电极装置。另外,将0.1m的高氯酸水溶液用作电解液。
在使用旋转环盘电极装置的耐久性试验(电位保持试验)的开始(寿命开始:bol)时,测量了碳催化剂的催化活性(初始催化活性)。
具体而言,通过使用具有包含碳催化剂的工作电极的三极旋转环盘电极装置进行循环伏安法(cv)。在cv中,首先,进行氮气鼓泡10分钟以除去电解液中的氧气。
然后,根据以下方案而进行cv测量。具体而言,在0.0v至1.0v的电位范围内(相对于普通氢电极(nhe))以50mv/秒的扫描速度进行五个循环(一个循环:1.0v→0.0v→1.0v)的电位扫描,并记录电流作为电位的函数。耐久性试验开始时的cv面积(bol-cv面积)(a·s)通过基于第五循环的所得伏安图的积分而得到。在所获得的伏安图中,将符号分配给数值,以使还原电流具有负值,而氧化电流具有正值。
作为上述伏安图的一个实例,通过使用具有包含实施例1中所得的碳催化剂的工作电极的旋转环盘电极装置而得到的第五循环时的伏安图示于图3中。在图3中,横轴表示电位(v相对于nhe),而纵轴表示电流(a)。在图3所示的伏安图中,由从1.0v至0.0v扫描施加电位时的曲线和从0.0v至1.0v扫描施加电位时的曲线而围成的阴影区域的面积通过积分作为cv面积(a·s)而获得。
此外,在cv测量之后,使电极以1600rpm的旋转速度旋转,并且当以20mv/秒的扫描速度进行电位扫描(bol-n2-lsv)时,记录下电流作为电位的函数。然后,进一步进行10分钟的氧气鼓泡,从而将饱和氧气填充到电解液中。然后,使电极以1600rpm的转速旋转,并且当以20mv/秒的扫描速度进行电位扫描(bol-o2-lsv)时,记录电流作为电位的函数。然后,从bol-o2-lsv中减去bol-n2-lsv,将纵轴的电流(ma)除以工作电极面积(cm2),从而得到以纵轴的氧还原伏安图,其表示电流密度(ma/cm2)。
作为氧还原伏安图的一个实例,在图4a和图4b的每一个中示出了通过使用具有包含实施例1中获得的碳催化剂的工作电极的旋转环盘电极装置而获得的氧还原伏安图。由图4a的虚线而包围的部分x以放大的方式示于图4b中。在图4a和图4b中,横轴表示电位(v相对于nhe),并且纵轴表示电流密度(ma/cm2)。
由图4a和图4b中的每一个所示的氧还原伏安图,作为表示碳催化剂的耐久性试验开始时的催化活性的指标,记录了还原电流为–10μa/cm2的电压(氧还原-起始电压bol-eo2)(v相对于nhe)和施加0.7v(相对于nhe)电压时的电流密度bol-i0.7(ma/cm2)。
然后,进行了耐久性试验(电位保持试验)。具体而言,用新鲜的0.1m高氯酸水溶液代替电解液,并进行10分钟的氧气鼓泡,从而用饱和氧填充电解液。然后,使电极以1600rpm的转速而旋转,并施加1.2v(相对于rhe)的电压。将该状态保持10分钟,并记录此期间的电流作为电位的函数。
然后,测量耐久性使用结束(寿命结束:eol)时碳催化剂的催化活性。具体而言,在不更换电解液的情况下进行氮气鼓泡10分钟,从而除去电解液中的氧气。
然后,以与耐久性测试开始时的测量相同的方式,依据以下方案而进行cv测量。具体而言,在0.0v至1.0v的电位范围内(相对于普通氢电极(nhe))以50mv/秒的扫描速度进行五个循环(一个循环:1.0v→0.0v→1.0v)的电位扫描,并记录电流作为电位的函数。
这里,耐久性试验结束时的cv面积(eol-cv面积)(a·s)通过基于第五循环的所得伏安图的积分而得到。此外,通过从eol-cv面积中减去bol-cv面积而计算cv面积增加量(a·s),并作为抗氧化性的指标而记录。
此外,在cv测量之后,使电极以1600rpm的旋转速度旋转,并且当以20mv/秒的扫描速度进行电位扫描(eol-n2-lsv)时,记录下电流密度作为电位的函数。然后,进一步进行10分钟的氧气鼓泡,从而将饱和氧气填充到电解液中。然后,使电极以1600rpm的转速旋转,并且当以20mv/秒的扫描速度进行电位扫描(eol-o2-lsv)时,记录电流密度作为电位的函数。然后,从bol-o2-lsv中减去bol-n2-lsv,将纵轴的电流(ma)除以工作电极面积(cm2),从而得到以纵轴的氧还原伏安图,其表示电流密度(ma/cm2)。
由上述得到的耐久性试验结束时的氧还原伏安图,记录了还原电流为–10μa/cm2的电压(氧还原-起始电压eol-eo2)(v相对于nhe)和施加0.7v(相对于nhe)电压时的电流密度eol-i0.7(ma/cm2)。然后,通过将耐久试验结束时的电流密度eol-i0.7(ma/cm2)除以耐久试验开始时的bol-i0.7(ma/cm2)所获得的值乘以100,从而计算出电流密度i0.7维持率(%)。
结果
通过以下对实施例1至17和比较例1至4中获得的碳催化剂进行评估而获得的结果示于图5:耐久性试验开始时的氧还原开始电位eo2(v)和电流密度i0.7(ma/cm2)表示初始催化活性;电流密度i0.7维持率(%)和cv面积增加量(a·s)表示耐久性;通过xrd而测量微晶尺寸lc(nm);通过tpd而测量co解吸量、co2解吸量和(co+co2)解吸量;通过拉曼光谱而测量的iv/ig比、d带半高宽(cm-1)和g带半高宽(cm-1);和bet比表面积(m2/g)。
如图5所示,在比较例1和3的碳催化剂中,耐久性试验开始时的氧还原开始电位eo2为0.827v至0.828v,电流密度i0.7为–1.71至–1.72。但是,在耐久性试验过程中,电流密度i0.7维持率为87.1%至87.8%,cv面积增加量为6.37×10-5a·s至6.42×10-5a·s。具体而言,在比较例1和3的碳催化剂中,耐久性试验开始时的催化活性较高,但是耐久性低。
同时,在比较例2和4的碳催化剂中,在耐久性试验过程中,电流密度i0.7维持率为92.9%至93.8%,并且cv面积增加量为5.81×10-5a·s至5.87×10-5a·s,但是氧还原开始电位eo2为0.803v至0.804v,并且在耐久性试验开始时的电流密度i0.7为–0.95至–0.97。具体而言,在比较例2和4的碳催化剂中,耐久性较高,但耐久性开始时的催化活性较低。
相反,在实施例1至17的碳催化剂中,在耐久性试验开始时,氧还原开始电位eo2为0.826v至0.837v,电流密度i0.7为–1.57至–1.80。另外,在耐久性试验过程中,在实施例1至17的碳催化剂中,电流密度i0.7维持率为90.6%至93.7%,cv面积增加量为4.74×10-5a·s至5.25×10-5a·s。特别而言,在耐久性试验过程中,在实施例2至17的碳催化剂中,电流密度i0.7维持率为91.8%至93.7%,并且cv面积增加量为4.74×10-5a·s至4.96×10-5a·s。
具体而言,实施例1至17的碳催化剂具有优异的催化活性和优异的耐久性两者的特征。特别是实施例2至17的碳催化剂的耐久性具有显著优异的耐久性。
此外,通过比较例1和3的碳催化剂的xrd测量的微晶尺寸lc为1.01nm以下,并且比较例2和4的碳催化剂的微晶尺寸lc为1.24nm以上。相反,通过实施例1至17的碳催化剂的xrd测量的微晶尺寸lc为0.98nm至1.13nm。
此外,通过比较例1至3的碳催化剂的tpd测量的co解吸量为2,150.0μmol/g以上,并且通过比较例2和4的碳催化剂的那些为552.0μmol/g以下。相反,通过实施例1至17的碳催化剂的tpd测量的co解吸量为542.5μmol/g以下。
此外,通过比较例1和3的碳催化剂的tpd测量的co2解吸量为200.0μmol/g以上,并且比较例2和4的碳催化剂的那些为101.4μmol/g以上。减。相反,实施例1至17的碳催化剂的通过tpd测量的co2解吸量为85.2μmol/g以下,实施例2至17的碳催化剂的那些为54.0μmol/g或更小。
此外,比较例1和3的碳催化剂通过tpd测量的(co+co2)解吸量为2,350.0μmol/g以上,比较例2和4的碳催化剂的那些为653.4μmol/g以下。相反,实施例1至17的碳催化剂通过tpd测得的(co+co2)解吸量为618.2μmol/g以下,实施例2至17的碳催化剂的那些为551.7μmol/g以下。
此外,比较例1至4的碳催化剂在拉曼光谱中的iv/ig比为0.50以上。相反,实施例1至17的碳催化剂在拉曼光谱中的iv/ig比为0.31至0.48。
此外,比较例1至4的碳催化剂的拉曼光谱中的d带半高宽为180cm-1以上。相反,实施例1至17的碳催化剂的拉曼光谱中的d带半高宽为179cm-1以下。
此外,比较例1至4的碳催化剂的拉曼光谱中的g带半高宽为81cm-1以上。相反,实施例1至17的碳催化剂的拉曼光谱中的g带半高宽为80cm-1以下。
此外,比较例1至4的碳催化剂的bet比表面积为1,332m2/g至1,371m2/g。相反,实施例1至17的碳催化剂的bet比表面积为1,232m2/g至1,680m2/g。
此外,尽管并未示出,但是实施例1至17的碳催化剂通过xps测得的n/c比各自为2.0%以上,通过元素分析测得的氮含量为2.3重量%以上,最大粒径为10.0μm以下,平均粒径为0.60μm以下。
锌空气电池的制造
制造具有在其中形成包含碳催化剂的催化剂层的电池电极。具体而言,首先,将0.25克实施例2的碳催化剂和电解质溶液装入样品瓶中,并在超声浴中处理10分钟。然后,将所得物用均化器以25,000rpm搅拌10分钟,并且进一步用超声均化器以30w的输出和20khz的频率处理10分钟,从而获得用于催化剂层的含有均匀分散于其中的碳催化剂的浆状组合物。
将所得的用于催化剂层的浆状组合物施加于面积为9cm2的气体扩散层上(sglcarbonco.,ltd制造的“29bc”)(3.0cm×3.0cm),以使电池电极的每单位面积的碳催化剂的含量为1.5mg/cm2,然后干燥,从而在气体扩散层上形成催化剂层。因此,得到具有含有形成于其中的碳催化剂的催化剂层的电池电极。此外,为了比较,以相同的方式获得了电池电极,不同之处在于,使用了高导电性的炭黑(lioncorporation制造的ketjen黑)来代替所述碳催化剂。
然后,制造包括如上所述而获得的电池电极的锌空气电池。具体而言,制备两个切成尺寸为8cm×6cm的铝层压板(由dainipponprintingco.,ltd.制造)。将铝层压板之一的一部分切割,以形成正方形的窗口部分(2cm×2cm)。
此外,制备了切成尺寸为3cm×9cm的镍板(厚度:0.1mm,由thenilacocorporation制造)。从镍板上切下尺寸为2cm×6cm的部分,从而获得包括正方形基部(3cm×3cm)和从基部延伸的矩形端部分(1cm×6cm)的l形镍板。
然后,将镍板堆叠在一个铝层压板上,使得镍板的基部从一个铝层压板的窗口部分露出。另外,在镍板的基部的一部分(2cm×2cm)中形成了作为空气入孔的规则排列的9个孔(直径均为3mm)(3个孔×3个孔),其从铝层压板的窗口部分露出。
然后,将电池电极堆叠在镍板的基部上,以使铝层压板相反侧上的镍板基部的表面与如上述获得的电池电极(3cm×3cm)的气体扩散层彼此接触。此外,布置了热焊接带(由dainipponprintingco.,ltd.制造),以便从电池电极表面的宽度为0.5cm的框状外周部分在铝层压板周围延伸,其中所述电池电极表面在镍板相反的一侧(即催化剂层的表面)。通过热熔接带的热熔接,将电池电极、镍板和铝层压板成为一体,从而得到正极(空气电极)。
同时,以如在镍板上相同方式从切成尺寸为3cm×9cm的铜箔(厚度:20μm,由hohsencorp.制造)而切出尺寸为2cm×6cm的部分,从而获得包括正方形基部(3cm×3cm)和从基部延伸的矩形端部分(1cm×6cm)的l形铜箔。然后,将铜箔的基部与切成3cm×3cm的尺寸的锌板(厚度:0.5mm,nilacocorporation制造)用超声焊接机而彼此焊接,从而获得锌负极。
然后,将切成尺寸为3cm×3cm的纤维素隔板(tf40-50,由nipponkodoshicorporation制造)堆叠在正极的催化剂层表面上。然后,将锌负电极堆叠在纤维素隔板上,以使正极相反侧上的纤维素隔板的表面与锌负电极的锌板表面彼此接触。此时,布置了镍板和铜箔,以使镍板的端部和铜线圈的端部并不彼此重叠。
此外,将另一个铝层压板(8cm×6cm)堆叠在纤维素隔板相反侧上的锌负极的表面(即铜箔的表面)上。然后,这对层叠的铝层压板的四个侧面中的三个侧面进行热焊接,从而形成在一侧开口的铝层压袋。
即,在所述铝层压袋中,l形镍板、电池电极的气体扩散层、电池电极的催化剂层、纤维素隔板、锌电极的锌板和锌电极的l形铜箔以所述顺序而从具有形成于其中的窗口部分的一个铝层压板到另一铝层压板而排列。
此外,通过铝层压袋的开口注入用作电解液的4mol/l的氢氧化钾水溶液(由hayashipurechemicalind.,ltd.制造)。最后,通过热焊接将铝层压袋的开口封闭,以获得锌空气电池。将延伸到电池外部的镍板的端部用作正极端,而铜箔的端部用作负极端。
制造镁空气电池
以与上述锌空气电极相同的方式而制造包括如上所述获得的电池电极的镁空气电池。具体而言,将包括一个正方形的基部(3cm×3cm)和一个从基部延伸的矩形端部(1cm×6cm)的l型镁合金板用作镁负极以代替上述锌负极,从而形成在一侧开口的铝层压袋,其中l形镍板、电池电极的气体扩散层、电池电极的催化剂层、纤维素隔板、镁负极以所述顺序而从具有形成于其中的窗口部分的一个铝层压板到另一铝层压板而排列。
然后,通过铝层压袋的开口注入用作电解液的4mol/l氯化钠水溶液(kantochemicalindustryco.,ltd.制备)。最后,通过热焊接封闭铝层压袋的开口,以获得镁空气电池。将延伸到电池外部的镍板的端部用作正极端,镁合金板的端部用作负极端。
空气电池的特性评价
通过使用充电/放电装置(hj0505sm8a,hokutodenkocorporation制造),以0.5v的电池电压设置为截止电压而对如上所述制造的锌空气电池和镁空气电池中的每一个的最大输出密度进行测量。
锌空气电池和镁空气电池中的每一个的正电极和负电极的构造以及通过测量其最大输出密度(mw/cm2)而获得的结果示于图6。如图6所示,在锌空气电池和镁空气电池中的任何一个中,与具有包含炭黑的正极的空气电池相比,在具有包含碳催化剂的正极的空气电池中获得了显著更高的最大输出密度。具体而言,证实了所述碳催化剂在空气电池中表现出优异的催化活性。
此外,对锌空气电池和镁空气电池各自的耐久性进行了评估。具体而言,对如上所述制造的空气电池进行了输出特性测试。然后,以10ma/cm2的电流值进行恒定电流放电,直到高至30%的理论值。然后,再次进行输出特性测试。然后,将通过恒定电流放电之后的输出值除以恒定电流放电之前的输出值而获得的值乘以100,从而计算输出维持率(%)。
锌空气电池和镁空气电池中的每一个的正电极和负电极的构造以及通过测量其输出维持率(%)而获得的结果示于图7。如图7所示,在锌空气电池和镁空气电池中的任何一个中,与具有包含炭黑的正极的空气电池相比,在具有包含碳催化剂的正极的空气电池中获得了显著更高的输出维持率。具体而言,证实了所述碳催化剂在空气电池中表现出优异的耐久性。
- 还没有人留言评论。精彩留言会获得点赞!