加氢催化剂、制备方法及在苯酚加氢制备环己酮的应用与流程
1.本发明涉及催化领域,尤其涉及加氢催化剂、制备方法及在苯酚加氢制备环己酮的应用。
背景技术:
2.环己酮是作为生产己内酰胺和己二酸的重要原料,同时也是重要的有机溶剂。目前工业上通常采用的工艺包括环己烷氧化法、环己醇脱氢制环己酮或苯酚一步加氢制环己酮。而苯酚加氢作为一种环境友好的生产工艺,越来越受到人们的关注。作为苯酚加氢制环己酮的催化剂,其不仅需要具备较高的加氢活性,同时还要有较高的环己酮选择性。
3.公开号为cn110563564a的中国发明专利申请公开了一种负载有钯的三氧化二氯催化剂。该催化剂应用于苯酚加氢制备环己酮的反应,以苯酚为原料,以环己酮为溶剂,明显会造成产能的降低,且如果应用于工业上的苯酚加氢催化反应,出于产能方面的考虑,生成的部分环己酮需要经分离提纯和冷却后再与原料苯酚混合预热,然后重新进入反应系统中。反复的提纯和升降温使能耗加大,反应系统效率低下。
4.因此,有必要开发一种新型的应用于苯酚加氢制备环己酮的加氢催化剂,以避免现有技术中存在的上述问题。
技术实现要素:
5.本发明的目的在于提供一种加氢催化剂、所述加氢催化剂的制备方法以及在苯酚加氢制备环己酮的应用,以获得良好的环己酮选择性和苯酚转化率。
6.为实现上述目的,本发明的加氢催化剂包括铝氧化物以及均负载于所述铝氧化物的钯、第一助成分和第二助成分;,所述第一助成分包含第一助金属元素,所述第二助成分包含第二助金属元素,所述第一助金属元素为碱土金属中的任意一种,所述第二助金属元素为
ⅵ
b族金属元素、ⅱb族金属元素或ⅲa族金属元素中的任意一种;以占所述铝氧化物的质量百分比计,所述钯的含量为0.2-1.0%,所述第一助金属元素的含量为1.0-3.0%,所述第二助金属元素的含量为0.2-0.6%。
7.本发明的所述加氢催化剂的有益效果在于:以钯作为主金属,包含碱土金属的第一助成分和包含
ⅵ
b族金属元素、ⅱb族金属元素或ⅲa族金属元素中的任意一种的第二助成分分别负载于铝氧化物,结合对各组分含量的调控,有利于活性中心的高度分散以及钯、所述第一助金属元素和所述第二助金属元素之间能够实现良好的协同效应,进而在应用于苯酚加氢制备环己酮的反应中获得良好的环己酮选择性和苯酚转化率。
8.优选的,所述第一助金属元素为钙和镁中的任意一种,所述第二助金属元素为锌、镓和铬中的任意一种。其有益效果在于:有利于不同金属组分之间的良好协同效应。
9.优选的,所述铝氧化物呈颗粒状,所述铝氧化物的粒度为1.0-5.0毫米,比表面积为80-300平方米/克,平均孔径为5-40纳米。
10.所述的加氢催化剂的制备方法包括:
11.s1:提供原始载体,在500-800℃下对所述原始载体进行3-8小时的焙烧后,使用包括含钯物质的浸渍液对所述预处理原始载体进行第一浸渍,以得到含钯原始载体,所述原始载体包括铝氧化物,所述含钯物质中的钯占所述原始载体的质量百分比为0.2%-1.0%;
12.s2:使用质量浓度为8-15%的无机碱溶液对所述含钯原始载体进行沉淀,然后对得到的沉淀物进行洗涤和干燥,以得到前驱体;
13.s3:使用包括第一助剂和第二助剂的混合溶液对所述前驱体进行第二浸渍后干燥,以得到负载有助剂的前驱体,所述第一助剂含有第一助金属元素,所述第二助剂含有第二助金属元素,所述第一助金属元素占所述原始载体的质量百分比为1-3%,所述第二助金属元素占所述原始载体的质量百分比为0.2-0.6%;
14.s4:使用由还原气体和保护气体组成的混合气体在150-300℃下对所述负载有助剂的前驱体进行3-8小时的还原,然后使用所述保护气体在350-500℃下对经所述还原得到的催化剂进行1.5-3小时的高温处理,以得到所述加氢催化剂。
15.本发明的所述制备方法的有益效果在于:通过所述第一浸渍、所述沉淀以及所述第二浸渍实现了钯、所述第一助金属元素和所述第二助金属元素的负载,结合调控所述浸渍液、所述无机碱溶液和所述混合溶液中的钯含量、无机碱浓度以及所述第一助金属元素和所述第二助金属元素的含量以及所述还原的工艺参数,实现了对所述加氢催化剂中各组分含量的调控,有利于活性中心的高度分散以及各金属组分之间能够实现良好的协同效应;另外,所述还原结束后进行的所述高温处理,能够加强钯、第一助金属元素和第二助金属元素分别与所述原始载体结合的牢固程度,有利于通过提高催化剂的稳定性来延长使用寿命。
16.优选的,所述步骤s3中,所述混合溶液的体积不小于所述原始载体的饱和吸水体积,且不大于所述原始载体的饱和吸水体积的1.5倍,所述第二浸渍的温度为20-50℃,浸渍时间为2-6小时,所述第一助剂为水溶性的碱土金属化合物,所述第二助剂为
ⅵ
b族金属元素、ⅱb族金属元素或ⅲa族金属元素中的任意一种的水溶性化合物。其有益效果在于:有利于所述第一助金属元素和所述第二助金属元素在所述原始载体表面的高度分散。
17.优选的,所述步骤s4中,每小时通入的所述还原气体的摩尔量为所述含钯物质中的钯的摩尔量的20-100倍,所述保护气体的摩尔量为所述还原气体摩尔量的1-3倍。其有益效果在于:有利于提高所述加氢催化剂的稳定性。
18.优选的,所述第一浸渍的温度为20-50℃,浸渍时间为3-16小时,所述浸渍液的质量为所述原始载体质量的1-4倍,所述含钯物质为水溶性的钯化合物。其有益效果在于:有利于钯在所述原始载体表面的高度分散。
19.优选的,所述步骤s2中,所述无机碱溶液的温度为20-50℃,所述沉淀的时间为2-6小时。其有益效果在于:有利于钯在所述原始载体表面的高度分散以及相互结合的牢固程度。
20.本发明的所述加氢催化剂在苯酚加氢制备环己酮的应用包括:所述加氢催化剂装填于固定床反应器,使所述固定床反应器内部充满保护气体,然后将熔融态苯酚、氢气和保护气体混合后进入所述固定床反应器内部并流经所述加氢催化剂,以通过一步加氢反应得到环己酮。
21.本发明的所述应用的有益效果在于:由于使用了所述加氢催化剂,所述加氢催化
剂具有良好的催化活性,使得苯酚加氢制备环己酮的反应无需使用溶剂,也无需对苯酚进行气化,而是以所述熔融态苯酚为原料就能够通过一步加氢反应制备环己酮,在提高产能的同时能够有效降低能耗。
22.优选的,所述熔融态苯酚的质量空速为0.2-0.8小时-1
,所述氢气的摩尔量为所述熔融态苯酚摩尔量的3-6倍,所述保护气体的摩尔量为所述氢气摩尔量的1-3倍,所述一步加氢反应的温度为120-160℃,所述固定床反应器内的表压为0-0.2兆帕,所述熔融态苯酚进入所述固定床反应器前的温度高于苯酚的熔点且不超过100℃。其有益效果在于:反应条件温和,能耗低,且有利于获得良好的环己酮选择性和苯酚转化率。
具体实施方式
23.为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。除非另外定义,此处使用的技术术语或者科学术语应当为本发明所属领域内具有一般技能的人士所理解的通常意义。本文中使用的“包括”等类似的词语意指出现该词前面的元件或者物件涵盖出现在该词后面列举的元件或者物件及其等同,而不排除其他元件或者物件。
24.针对现有技术存在的问题,本发明实施例提供了一种应用于苯酚加氢制备环己酮的加氢催化剂,以获得良好的环己酮选择性和苯酚转化率。
25.本发明实施例中,粒度定义为颗粒状物质的单轴最大尺寸。比表面积和平均孔径由bet比表面积分析得到,具体的分析方法为本领域技术人员的常规技术手段,在此不做赘述。
26.本发明一些实施例中,所述加氢催化剂能够应用于苯酚在无溶剂存在下通过一步加氢制备环己酮的反应,即原料为熔融态苯酚和氢气。
27.本发明实施例中,所述加氢催化剂包括铝氧化物以及均负载于所述铝氧化物的钯、第一助成分和第二助成分。所述钯为主金属,所述铝氧化物为载体。
28.本发明一些实施例中,所述加氢催化剂由所述铝氧化物以及负载于所述铝氧化物的所述钯、所述第一助成分和所述第二助成分组成。
29.具体的,所述第一助成分包含第一助金属元素,所述第二助成分包含第二助金属元素,以占所述铝氧化物的质量百分比计,所述钯的含量为0.2-1.0%,所述第一助金属元素的含量为1.0-3.0%,所述第二助金属元素的含量为0.2-0.6%。
30.本发明一些实施例中,所述第一助金属元素为碱土金属中的任意一种,所述第二助金属元素为
ⅵ
b族金属元素、ⅱb族金属元素或ⅲa族金属元素中的任意一种。
31.本发明一些实施例中,所述第一助金属元素为钙和镁中的任意一种,所述第二助金属元素为锌、镓和铬中的任意一种,所述铝氧化物为三氧化二铝,以有利于不同金属组分之间的良好协同效应。
32.本发明一些实施例中,所述第一助成分为所述第一助金属元素的氧化物,所述第二助成分为所述第二助金属元素的氧化物。
33.本发明一些实施例中,所述第一助成分为氧化钙和氧化镁中的任意一种,所述第
二助成分为氧化锌、三氧化二镓和三氧化二铬中的任意一种。
34.本发明一些实施例中,所述加氢催化剂中作为载体的铝氧化物呈颗粒状,所述铝氧化物的粒度为1.0-5.0毫米,比表面积为80-300平方米/克,平均孔径为5-40纳米。
35.本发明实施例还提供了所述加氢催化剂的制备方法,包括步骤s1、步骤s2、步骤s3和步骤s4。
36.本发明实施例的所述步骤s1包括:提供原始载体,在500-800℃下对所述原始载体进行3-8小时的焙烧,以形成稳定的孔结构。所述原始载体包括铝氧化物。
37.本发明一些实施例中,所述原始载体的比表面积为80-300平方米/克,平均孔径为5-40纳米。
38.本发明一些实施例中,所述原始载体呈颗粒状,所述颗粒状包括条形、球形和圆柱形,所述原始载体的粒度为1-5毫米。
39.本发明实施例1-5中,除实施例3的原始载体为柱形,其余实施例的原始载体均为球形。
40.本发明实施例1-5的所述步骤s1中,所述原始载体均由三氧化二铝组成,且质量均为50克。所述原始载体的比表面积bet、粒度d、平均孔径d
ape
、所述焙烧的温度t
roa
和时间t
roa
请参见表1。
41.表1
42.实施例编号12345bet/平方米.克-1
20030028080150d/毫米23521d
ape
/纳米215124020t
roa
/℃500800600500700t
roa
/小时63845
43.本发明实施例的所述步骤s1还包括:使用包括含钯物质的浸渍液对所述预处理原始载体进行第一浸渍,以得到含钯原始载体。其中,所述第一浸渍的温度为20-50℃,浸渍时间为3-16小时,以有利于钯在所述原始载体表面的高度分散。所述第一浸渍的温度具体为所述浸渍液的温度。
44.具体的,所述焙烧结束后,待所述预处理原始载体冷却至室温,再进行所述第一浸渍。
45.具体的,将所述预处理原始载体浸没于所述浸渍液,以进行所述第一浸渍。
46.本发明一些实施例中,所述含钯物质为水溶性的钯化合物,所述浸渍液为所述含钯物质的水溶液。
47.具体的,所述含钯物质中的钯占所述原始载体的质量百分比为0.2%-1.0%,所述浸渍液的质量为所述原始载体的1-4倍。
48.具体的,所述钯化合物为氯化钯、硝酸钯和醋酸钯中的任意一种。
49.本发明实施例1-5的所述步骤s1中,所述浸渍液为钯化合物的水溶液,实施例1和实施例2的钯化合物为氯化钯,实施例3和实施例5的钯化合物为硝酸钯,实施例4的钯化合物为醋酸钯。各实施例的浸渍液的质量m
pdc
、所述浸渍液中钯的质量m
pd
、所述浸渍液的温度t
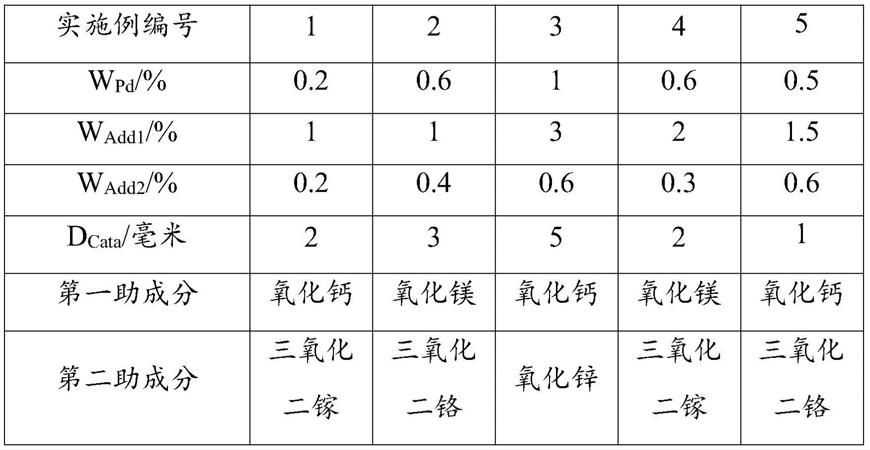
lpd
以及浸渍时间t
lpd
请参见表2。
50.表2
51.实施例编号12345m
pdc
/克50200150100100m
pd
/克0.10.30.50.30.25t
lpd
/℃2050305040t
lpd
/小时16121238
52.本发明实施例的步骤s2包括:使用质量浓度为8-15%的无机碱溶液对所述含钯原始载体进行沉淀,然后对得到的沉淀物进行洗涤和干燥,以得到前驱体。
53.通过所述沉淀能够促使钯化合物在所述原始载体晶粒表面的扩散和表面反应,通过所述洗涤去除杂离子,防止杂质吸附在所述原始载体表面而影响得到的加氢催化剂的催化性能。通过所述干燥去除游离水和吸附水。
54.具体的,所述沉淀为静置沉淀,所述沉淀的温度,即所述无机碱溶液的温度为20-50℃,所述沉淀的时间为2-6小时,以有利于钯在所述原始载体表面的高度分散以及相互结合的牢固程度。
55.本发明一些实施例中,所述无机碱溶液中的溶质为氢氧化钠、碳酸钠、碳酸氢钠、氢氧化钾、碳酸氢铵和氨水中的一种,溶剂为水。
56.本发明实施例1-5的所述步骤s2中,所述洗涤具体为:使用去离子水对所述沉淀物进行洗涤,直至收集到的滤液的ph值为7且不含有氯离子。所述干燥在常压下进行,所述干燥的温度为110℃。无机碱的种类、无机碱水溶液的质量m
as
、无机碱水溶液中的无机碱的质量浓度w
a
、所述无机碱水溶液的温度t
p
以及所述沉淀的时间t
p
请参见表3。
57.表3
58.实施例编号12345无机碱碳酸钠碳酸氢钠氢氧化钠碳酸氢铵氢氧化钾m
as
/克12.5152012.515w
a
/%81081512t
p
/℃5020304050t
p
/小时26423
59.本发明实施例的所述步骤s3包括:使用包括第一助剂和第二助剂的混合溶液对所述前驱体进行第二浸渍后干燥,以得到负载有助剂的前驱体。
60.具体的,所述第一助剂含有第一助金属元素,所述第二助剂含有第二助金属元素,以使所述第一助金属元素和所述第二助金属元素同步负载于所述前驱体。
61.所述步骤s3中的干燥用于去除吸附水和游离水。
62.本发明一些实施例中,所述第一助剂为水溶性的碱土金属化合物,所述第二助剂为
ⅵ
b族金属元素、ⅱb族金属元素或ⅲa族金属元素中的任意一种的水溶性化合物。所述第一助剂的水溶液与所述第二助剂的水溶液之间不发生沉淀反应。
63.具体的,所述第一助剂为水溶性钙盐和水溶性镁盐中的任意一种,所述第二助剂为水溶性镓盐、水溶性锌盐和水溶性铬盐中的任意一种。
64.更具体的,所述第一助剂为钙的硝酸盐和镁的硝酸盐中的任意一种,所述第二助剂为镓的硝酸盐、锌的硝酸盐和铬的硝酸盐中的任意一种。
65.本发明一些实施例的所述步骤s3中,所述第一助金属元素占所述原始载体的质量百分比为1-3%,所述第二助金属元素占所述原始载体的质量百分比为0.2-0.6%。所述混合溶液的体积不小于所述原始载体的饱和吸水体积,且不大于所述原始载体的饱和吸水体积的1.5倍,以对所述前驱体进行浸渍,使得所述混合溶液充满所述前驱体的孔结构,且轻拿所述前驱体悬空后不会有所述混合溶液滴落。
66.具体的,所述原始载体的饱和吸水体积的测定方法为:用超声波清洗所述原始载体以去除表面的杂质后在110℃下烘干至恒重,得到干态重量,然后将所述原始载体放置于清洁的容器中,对容器抽真空以使容器内部压力不高于27帕;抽真空结束后静置5分钟,然后向容器中缓慢注入去离子水以浸没所述原始载体,注入去离子水的过程中使容器内部的压力始终不高于27帕;所述去离子水注入完毕后静置5分钟,取出吸水的原始载体;使用被去离子水浸润的无纺布去除所述吸水的原始载体外表面的游离水后对得到的原始载体称重,得到湿态重量。以所述湿态重量与所述干态重量的差值作为所述原始载体的饱和吸水体积。
67.本发明一些实施例中,所述第二浸渍的温度为20-50℃,浸渍时间为2-6小时,以有利于所述第一助金属元素和所述第二助金属元素在所述原始载体表面的高度分散。
68.本发明实施例1-5的所述步骤s3中,第一助剂和第二助剂的种类、第一助剂中的第一助金属元素的含量m
ad1
、第二助剂中的第二助金属元素的含量m
ad2
、所述第二浸渍的温度t
ad
和时间t
ad
,以及所述干燥的温度t
d2
和时间t
d2
请参见表4。
69.表4
70.实施例编号12345第一助剂硝酸钙硝酸镁硝酸钙硝酸镁硝酸钙第二助剂硝酸镓硝酸铬硝酸锌硝酸镓硝酸铬m
ad1
/克0.50.51.510.75m
ad2
/克0.10.20.30.150.3t
ad
/℃2050403050t
ad
/小时62365t
d2
/℃150300200250300t
d2
/小时42343
71.本发明实施例的所述步骤s4包括:使用由还原气体和保护气体组成的混合气体在150-300℃下对所述负载有助剂的前驱体进行3-8小时的还原。
72.本发明一些实施例中,每小时通入的所述还原气体的摩尔量为所述含钯物质中的钯的摩尔量的20-100倍,所述保护气体的摩尔量为所述还原气体摩尔量的1-3倍,以有利于提高所述加氢催化剂的稳定性。
73.本发明一些实施例的所述步骤s4中,所述还原气体为氢气,所述保护气体为氮气或惰性气体,所述惰性气体为氦气、氖气、氩气、氪气、氙气和氡气中的一种或多种。
74.本发明实施例1-5的所述步骤s4中,所述还原气体为氢气,所述保护气体为氮气。将一定流量的氢气与一定流量的氮气混合后使混合气体通过放置有所述负载有助剂的前驱体的床层,以进行所述还原。所述氢气的流量v
h2
、所述氮气的流量v
n2
以及所述还原的温度t
red
和时间t
red
请参见表5。
75.表5
76.实施例编号12345v
h2
/毫升.分钟-1
71051054244v
n2
/毫升.分钟-1
211053008488t
red
/℃180250150200200t
red
/小时34886
77.本发明实施例的所述步骤s4还包括:所述还原完毕后,使用所述保护气体在350-500℃下对经所述还原得到的催化剂进行1.5-3小时的高温处理,以得到所述加氢催化剂。通过所述高温处理能够加强钯、所述第一助金属元素和所述第二助金属元素分别与所述原始载体结合的牢固程度,有利于通过提高催化剂的稳定性来延长使用寿命。
78.本发明实施例1-5的所述步骤s4中,所述还原完毕后,切断所述氢气的气流,在所述氮气的气氛下进行所述高温处理。所述高温处理的温度t
h
和时间t
h
请参见表6。
79.表6
80.实施例编号12345t
h
/℃350500400500400t
h
/小时31.5223
81.本发明实施例1-5得到的加氢催化剂的粒度d
cata
,以及主金属钯的含量w
pd
、第一助成分的种类和所述第一助成分中的第一助金属元素的含量w
add1
以及第二助成分的种类和所述第二助成分中的第二助金属元素的含量w
add2
请参见表7。各个实施例的加氢催化剂的比表面积请参见表1中对应实施例的原始载体的比表面积。表7中的含量指占所述加氢催化剂中的三氧化二铝载体的质量百分比。
82.表7
[0083][0084]
本发明的所述加氢催化剂在苯酚加氢制备环己酮的应用包括:所述加氢催化剂装填于固定床反应器,使所述固定床反应器内部充满保护气体,然后将熔融态苯酚、氢气和保护气体混合后进入所述固定床反应器内部并流经所述加氢催化剂,以通过一步加氢反应得到环己酮。由于使用了所述加氢催化剂,所述加氢催化剂具有良好的催化活性,使得苯酚加
氢制备环己酮的反应无需使用溶剂,也无需对苯酚进行气化,而是以所述熔融态苯酚为原料就能够通过一步加氢反应制备环己酮,在提高产能的同时能够有效降低能耗。
[0085]
本发明一些实施例中,所述熔融态苯酚的质量空速为0.2-0.8小时-1
,所述氢气的摩尔量为所述熔融态苯酚摩尔量的3-6倍,所述保护气体的摩尔量为所述氢气摩尔量的1-3倍,所述一步加氢反应的温度为120-160℃,所述固定床反应器内的表压为0-0.2兆帕,所述熔融态苯酚进入所述固定床反应器前的温度高于苯酚的熔点且不超过100℃。可见,所述一步加氢反应的反应条件温和,能耗低。
[0086]
具体的,所述表压指所述固定床反应器内部的测试压力与大气压之间的压力差。
[0087]
本发明实施例还提供了对比例1和对比例2。
[0088]
对比例1提供了第一对比催化剂,所述第一对比催化剂为负载有钯的三氧化二铝催化剂,钯占所述三氧化二铝的质量百分比为0.6%。所述第一对比催化剂的制备方法与实施例2的加氢催化剂的制备方法的区别在于:不具有所述步骤s1中的所述焙烧,不具有所述步骤s3,以及不具有所述高温处理。
[0089]
对比例2以上海盛邦化工有限公司生产的型号为fpmc-1001的负载有钯的氧化铝催化剂为第二对比催化剂。其中钯占所述氧化铝的质量百分比为1%。
[0090]
本发明实施例将实施例1-5的加氢催化剂、对比例1的所述第一对比催化剂和对比例2的所述第二对比催化剂各50克分别应用于苯酚一步加氢制备环己酮的反应。
[0091]
具体的,将待考察催化剂装填于固定床反应器,使用氮气置换反应系统内的空气;将苯酚在60℃下熔融以形成熔融态苯酚,并与氢气和所述氮气混合后从所述固定床反应器的顶部注入,以流经放置所述待考察催化剂的床层后从所述固定床反应器的底部流出,进行一步加氢反应。控制熔融态苯酚的质量空速为0.4小时-1
,氢气流量为300毫升/分钟,氮气的流量为450ml/min,反应温度为130℃,所述固定床反应器内的表压为0.01兆帕。对所述一步加氢反应得到的加氢产品进行气相色谱分析,得到苯酚的转化率以及环己酮的选择性。具体数值请参见表8。气相色谱分析的具体方法为本领域技术人员的常规技术手段,在此不做赘述。
[0092]
表8
[0093]
实施例苯酚转化率(%)环己酮选择性(%)实施例198.696.2实施例299.195.2实施例3100.093.8实施例499.694.2实施例5100.092.6对比例179.127.4对比例298.946.9
[0094]
参照表8,使用本发明实施例的加氢催化剂应用于苯酚一步加氢制备环己酮,苯酚的转化率在98%以上,最高可达100%,而环己酮的选择性不低于92%,最高可达96.2%,相比对比例1和2的两种对比催化剂具有更好的催化效果。
[0095]
本发明实施例以实施例2、3和4的加氢催化剂为例,调节所述一步加氢反应的工艺条件,考察苯酚的转化率以及环己酮的选择性。所述一步加氢反应的具体操作方法请参见
前述,熔融态苯酚的质量空速v1,氢气的流量v2,氮气的流量v3,反应温度t、所述固定床反应器内的表压p、苯酚转化率w1和环己酮选择性w2请参见表9。
[0096]
表9
[0097]
实施例编号234v1/小时-1
0.20.80.3v2/毫升.分钟-1
238476238v3/毫升.分钟-1
238953715t/℃120160140p/兆帕0.10.010.03w1/%97.098.599.6w2%99.097.793.2
[0098]
虽然在上文中详细说明了本发明的实施方式,但是对于本领域的技术人员来说显而易见的是,能够对这些实施方式进行各种修改和变化。但是,应理解,这种修改和变化都属于权利要求书中所述的本发明的范围和精神之内。而且,在此说明的本发明可有其它的实施方式,并且可通过多种方式实施或实现。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1