金属负载的催化剂、电池电极和电池的制作方法
1.本发明涉及金属负载的催化剂、电池电极和电池。
背景技术:
2.在专利文献1中,描述了一种用于空气电极的电极催化剂,该空气电极具有以下配置:(1)用于空气电极的电极催化剂含有由pt合金形成的第一催化剂颗粒和由平均粒径小于第一催化剂颗粒的平均粒径的纯pt形成的第二催化剂颗粒;和(2)pt合金具有由ptxm(1≤x≤4,m表示贱金属元素)表示的原子组成比。
3.在专利文献2中,描述了一种电极催化剂材料,其含有(i)含有多个单独的负载颗粒或聚集体的负载材料,(ii)含有第一金属和合金金属的第一颗粒,和(iii)由为铂或铱的第二金属或第二金属的氧化物形成的第二颗粒,其特征在于,单独的负载颗粒或聚集体每个具有分散在其上的第一颗粒和第二颗粒,并且第二颗粒的平均粒径小于第一颗粒的平均粒径。
4.引文列表
5.专利文献
6.[ptl 1]jp 2018-181739 a
[0007]
[ptl 2]jp 2019-517110 a
技术实现要素:
[0008]
技术问题
[0009]
然而,迄今为止,难以获得同时具有优异催化活性和耐久性的负载催化剂金属诸如铂的电极催化剂。
[0010]
本发明是鉴于上述问题而作出的,并且本发明的目的在于提供各自均具有优异的催化活性和耐久性的金属负载的催化剂、电池电极和电池。
[0011]
解决问题的技术方案
[0012]
为了实现上述目的,根据本发明的一个实施方案,提供了一种金属负载的催化剂,包括:碳载体;和催化剂金属颗粒,每个颗粒均包含负载在所述碳载体上的贵金属,其中每单位重量所述碳载体的第一孔的体积为0.20(cm3/g-载体)或更大,所述第一孔每个具有0.5nm或更大且2.0nm或更小的直径,其中每单位重量所述碳载体的第二孔的体积为0.20(cm3/g-载体)或更大,所述第二孔每个具有大于2.0nm且4.0nm或更小的直径,并且其中通过x射线光电子能谱法测定的贵金属的含量(重量%)与通过电感耦合等离子体质谱法测定的贵金属的含量(重量%)的比率为0.35或更大且0.75或更小。根据本发明的一个实施方案,提供了一种同时具有优异催化活性和耐久性的金属负载的催化剂。
[0013]
在金属负载的催化剂中,每单位重量所述碳载体的第三孔的体积为0.25(cm3/g载体)或更大,所述第三孔每个具有大于4.0nm且50.0nm或更小的直径。在金属负载的催化剂中,碳载体的每单位重量的每个具有大于4.0nm且50.0nm或更小的直径的第三孔的体积可
以为1.00(cm3/g载体)或更小。在金属负载的催化剂中,碳载体的每单位重量的每个具有大于4.0nm且50.0nm或更小的直径的第三孔的体积与每个具有大于2.0nm且4.0nm或更小的直径的第二孔的体积的比率可以为3.00或更小。
[0014]
在金属负载的催化剂中,使用通过分离在由cukα射线得到的粉末x射线衍射图中衍射角2θ在40
°
附近的衍射线而获得的一个或多个衍射峰的衍射角和半峰全宽由谢乐方程计算出的所述催化剂金属颗粒的平均微晶直径为7.0nm或更小。
[0015]
在金属负载的催化剂中,通过x射线光电子能谱法测量的氮原子浓度与碳原子浓度的比率可以为0.010或更大。在金属负载的催化剂中,通过将bet比表面积乘以金属负载的催化剂的重量与碳载体的重量之比而计算出的归一化比表面积可以为1,100(m2/g-载体)或更大。在金属负载的催化剂中,催化剂金属颗粒可以是铂颗粒。
[0016]
在金属负载的催化剂中,通过将氢吸附电量除以相对于铂的氢吸附的理论面积等效电量和负载在所述金属负载的催化剂上的铂的重量而获得的电化学表面积(h
2-ecsa)为20.0m2/g-铂或更大,在使用含有所述金属负载的催化剂的旋转圆盘电极的循环伏安法中测量所述氢吸附电量,并且其中通过将一氧化碳吸附电量除以相对于铂的一氧化碳吸附的理论面积等效电量和负载在所述金属负载的催化剂上的铂的重量而获得的电化学表面积(co-ecsa)为20.0m2/g-铂或更大,在使用含有所述金属负载的催化剂的旋转圆盘电极的溶出伏安法中测量所述一氧化碳吸附电量。
[0017]
金属负载的催化剂可具有碳结构,该所述碳结构在通过拉曼光谱法获得的拉曼光谱中表现出160cm-1
或更小的d带的半峰全宽,所述d带在1,360cm-1
附近具有峰顶。金属负载的催化剂可具有碳结构,该碳结构在通过拉曼光谱法获得的拉曼光谱中表现出90cm-1
或更小的g带的半峰全宽,所述g带在1,600cm-1
附近具有峰顶。金属负载的催化剂可具有碳结构,该碳结构表现出0.20或更大且0.50或更小的在通过拉曼光谱法获得的拉曼光谱中,在1,600cm-1
附近具有峰顶的g带和在1,360cm-1
附近具有峰顶的d带之间的最小强度与g带的强度的比率。
[0018]
金属负载的催化剂可以具有碳结构,该碳结构在程序升温脱附法中表现出1.20
×
10-5
(mol/g-载体)或更大的碳载体的每单位重量的从600℃到1,000℃的氮脱附量。金属负载的催化剂可以具有碳结构,该碳结构在程序升温脱附法中每单位重量所述碳载体从800℃到1,000℃表现出0.75
×
10-5
(mol/g-载体)或更大的氮脱附量。
[0019]
为了实现上述目的,根据本发明的一个实施方案,提供了一种包括上述金属负载的催化剂中的任一种的电池电极。根据本发明的一个实施方案,提供了一种同时具有优异催化活性和耐久性的电池电极。
[0020]
为了实现上述目的,根据本发明的一个实施方案,提供了一种包括电池电极的电池。根据本发明的一个实施方案,提供了一种同时具有优异催化活性和耐久性的电池。
[0021]
发明的有益效果
[0022]
根据本发明,提供了各自均具有优异催化活性和耐久性的金属负载的催化剂、电池电极和电池。
附图说明
[0023]
图1a是示出了粉末x射线衍射图中的峰分离的示例的说明图。
[0024]
图1b是示出了粉末x射线衍射图中的峰分离的示例的说明图。
[0025]
图2是示出了粉末x射线衍射图中的峰分离的结果的示例的说明图。
[0026]
图3是示出了透射电子显微镜图像中的催化剂金属颗粒的粒径的评估的示例的说明图。
[0027]
图4a是示出了在根据本发明的一个实施方案的实施例中获得的haadf-stem图像的示例的说明图。
[0028]
图4b是示出了在根据本发明的一个实施方案的实施例中获得的stem二次电子图像的示例的说明图。
[0029]
图5是示出了通过拉曼光谱法获得的拉曼光谱的评估的示例的说明图。
[0030]
图6a是示出了在根据本发明的一个实施方案的实施例中的金属负载的催化剂的特性的评估结果的示例的说明图。
[0031]
图6b是示出了在根据本发明的一个实施方案的实施例中的金属负载的催化剂的特性的评估结果的另一示例的说明图。
[0032]
图6c是示出了在根据本发明的一个实施方案的实施例中的金属负载的催化剂的特性的评估结果的又一示例的说明图。
具体实施方式
[0033]
现在,将描述根据本发明的一个实施方案的金属负载的催化剂(以下称为“本发明的催化剂”)、根据本发明的一个实施方案的电池电极(以下称为“本发明的电极”),和根据本发明的一个实施方案的电池(以下称为“本发明的电池”)。本发明不限于这些实施方案中描述的示例。
[0034]
本发明的催化剂是金属负载的催化剂,包括:碳载体;和催化剂金属颗粒,每个均包含负载在碳载体上的贵金属,其中碳载体的每单位重量的每个具有0.5nm或更大且2.0nm或更小的直径的第一孔的体积为0.20(cm3/g-载体)或更大,其中碳载体的每单位重量的每个具有大于2.0nm且4.0nm或更小的直径的第二孔的体积为0.20(cm3/g-载体)或更大,并且其中通过x射线光电子能谱法(xps)测定的贵金属的含量(重量%)与通过电感耦合等离子体质谱法(icp-ms)测定的贵金属的含量(重量%)的比率为0.35或更大且0.75或更小。
[0035]
本发明的催化剂中所含的碳载体是主要由碳形成的碳材料。碳载体中碳原子的含量可以为例如70重量%或更大、75重量%或更大、80重量%或更大、或85重量%或更大。
[0036]
碳载体可以是碳化材料。即,碳载体可以是通过将包含有机物质的原料碳化而获得的碳化材料。此外,碳载体可以是通过将包含有机物质和金属的原料碳化而获得的碳化材料。在这种情况下,金属优选为过渡金属。过渡金属可以是除铂之外的过渡金属,或者可以是除贵金属(例如钌(ru)、钯(pd)、铑(rh)、银(ag)、锇(os)、铱(ir)、铂(pt)和金(au))之外的过渡金属。
[0037]
碳载体可以在其骨架内部包含过渡金属。即,碳载体可以在形成其多孔结构的骨架内部包含过渡金属。具体地,当碳载体是通过将包含有机物质和过渡金属的原料碳化而生产的碳化材料时,碳载体包含源自在其生产时用于碳化的原料的过渡金属。即,碳载体在其骨架内部含有过渡金属,因为过渡金属包含在用于碳化的原料中。即使当碳载体通过金属去除处理生产时,源自原料的痕量过渡金属仍残留在碳载体内部。
[0038]
通过切割骨架并分析通过切割暴露的横截面来检测碳载体骨架内部的过渡金属。即,在碳载体为颗粒形式的情况下,当碳载体的颗粒被切割时,在通过切割暴露的颗粒的横截面中检测过渡金属。
[0039]
碳载体中所含的过渡金属可以通过例如icp-ms检测。碳载体的通过icp-ms的过渡金属含量计算为过渡金属原子的重量相对于碳载体的总重量的比例(重量%)。
[0040]
碳载体可以在其骨架内部含有0.01重量%或更多的过渡金属(例如,源自用于碳化的原料的过渡金属),或者可以含有0.02重量%或更多的过渡金属。此外,碳载体中过渡金属的含量可以为15重量%或更少,或者可以为10重量%或更少。碳载体中的过渡金属的含量可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0041]
源自用于碳化的原料的碳载体中所含的过渡金属可以是元素周期表中第iii-xii族的过渡金属,优选是元素周期表中第iii-xii族第四周期的过渡金属。
[0042]
具体地,碳载体中所含的过渡金属可以是例如选自由钪(sc)、钛(ti)、钒(v)、铬(cr)、锰(mn)、铁(fe)、钴(co)、镍(ni)、铜(cu)、锌(zn)、钇(y)、锆(zr)、铌(nb)、钼(mo)、钌(ru)、铑(rh)、钯(pd)、银(ag)、镧系元素(例如钆(gd))和锕系元素组成的组的一种或更多种或者两种或更多种,并且优选为选自由fe、co、ni、cu和zn组成的组的一种或更多种或者两种或更多种,特别优选选自由fe、co、ni和zn组成的组的一种或更多种或两种或更多种。
[0043]
碳载体可以是碳催化剂。在这种情况下,碳载体本身独立地表现出催化活性。具体地,碳载体可以表现出例如作为催化活性的还原活性和/或氧化活性,并且优选表现出氧还原活性和/或氢氧化活性,并且特别优选地表现出至少氧还原活性。
[0044]
碳载体可以是在其骨架内部含有过渡金属的碳催化剂。即,当碳载体是通过将包含有机物质和金属的原料碳化而获得的碳催化剂时,碳催化剂的碳结构包含金属。在这种情况下,认为碳催化剂的催化活性主要是由碳化形成的碳结构中包含的活性位点引起的,而不是源自用于碳化的原料的金属。前述内容由以下事实支持:即使当将包含源自用于碳化的原料的金属的碳催化剂进行金属去除处理以降低金属含量时,金属去除处理之后的碳催化剂的催化活性与金属去除处理之前的催化活性相比没有明显降低;与碳催化剂不同,通过将包含有机物质但不含金属的原料进行碳化获得的碳化材料的表面上负载金属而获得的碳材料在碳化后不具有优异的催化活性。
[0045]
本发明的催化剂中的待负载在碳载体上的催化剂金属颗粒没有特别限制,只要该催化剂金属颗粒是各自包含贵金属并表现出催化活性的金属颗粒即可,但优选是贵金属颗粒。贵金属颗粒例如优选为各自包含选自由钌(ru)、钯(pd)、铑(rh)、银(ag)、锇(os)、铱(ir)、铂(pt)和金(au)组成的组的一种或更多种的贵金属颗粒,更优选各自包含选自由ru、pd、rh、ir和pt组成的组的一种或更多种的贵金属颗粒,特别优选pt颗粒。
[0046]
贵金属颗粒为纯贵金属颗粒和/或贵金属合金颗粒。贵金属合金颗粒各自由贵金属(例如,选自由钌(ru)、钯(pd)、铑(rh)、银(ag)、锇(os)、铱(ir)、铂(pt)和金(au)组成的组的一种或更多种,优选选自由ru、pd、rh、ir和pt组成的组的一种或更多种,特别优选pt和一种或更多种非贵金属形成。
[0047]
非贵金属是除贵金属之外的金属并且没有特别限制,只要该非贵金属与贵金属形成合金即可,但优选是过渡金属。即,贵金属合金颗粒也可以包含贵金属和过渡金属的贵金属合金。在这种情况下,贵金属合金可以是贵金属和一种过渡金属的合金,或者可以是贵金
属和两种或更多种过渡金属的合金。
[0048]
具体地,贵金属合金优选为贵金属与选自由cu、mn、ce、au、pd、ru、nb、ti、fe、co和ni组成的组的一种或更多种过渡金属的合金,更优选贵金属与选自由fe、co和ni组成的组的一种或更多种过渡金属的合金,特别优选贵金属与选自由co和ni组成的组的一种或更多种过渡金属的合金。
[0049]
本发明的催化剂可以仅包含纯贵金属颗粒作为贵金属颗粒,可以仅包含贵金属合金颗粒,或者可以包含纯贵金属颗粒和贵金属合金颗粒,但优选包含至少纯贵金属。
[0050]
包括纯贵金属颗粒和/或贵金属合金颗粒的贵金属颗粒例如是各自包含由ru、pd、rh、ag、os、ir、pt和au组成的组的一种或更多种的贵金属颗粒,更优选各自包含选自由ru、pd、rh、ir和pt组成的组的一种或更多种的贵金属颗粒,特别优选各自包含pt的贵金属颗粒(pt颗粒)。
[0051]
具体地,例如,pt颗粒为纯pt颗粒和/或pt合金颗粒。纯pt显示出在通过x射线衍射(xrd)获得的xrd衍射图中在衍射角(2θ)为39.6
°
或更大且小于39.8
°
的位置处具有峰顶的衍射线。pt合金显示出在xrd衍射图中在衍射角(2θ)为39.9
°
或更大且小于43.0
°
的位置处具有峰顶的衍射线。
[0052]
pt合金是pt与一种或多种其他金属(除pt之外的金属:非pt金属)的合金。非pt金属没有特别限制,只要该非pt金属与pt形成合金即可,但优选是过渡金属。即,pt合金颗粒可以各自包含铂与过渡金属的铂合金。在这种情况下,pt合金可以是pt与一种过渡金属的合金,或者可以是pt与两种或更多种过渡金属的合金。
[0053]
具体地,pt合金优选为pt与选自由cu、mn、ce、au、pd、ru、nb、ti、fe、co和ni组成的组的一种或更多种过渡金属的合金,更优选pt与选自由fe、co和ni组成的组的一种或更多种过渡金属的合金,特别优选pt与选自由co和ni组成的组的一种或更多种过渡金属的合金。
[0054]
本发明的催化剂可以仅包含纯pt颗粒作为pt颗粒,可以仅包含pt合金颗粒,或者可以包含纯pt颗粒和pt合金颗粒,但优选包含至少纯pt。
[0055]
即,本发明的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为例如优选5%或更大,特别优选10%或更大。
[0056]
在本发明的催化剂中包含pt和过渡金属的pt合金颗粒有助于催化活性。当pt与贱金属形成合金时,在晶格中产生应变,并且提高了其氧还原催化活性。上述各自含有过渡金属的pt合金颗粒通过具有在其中产生适当应变的晶格而各自表现出优异的催化活性。
[0057]
通过icp-ms测量获得的本发明的催化剂中的贵金属(例如pt)的含量(负载在金属负载的催化剂上的催化剂金属颗粒中所含的贵金属的重量相对于金属负载的催化剂的重量的比例)可以为例如1.0重量%或更大,并且优选为5.0重量%或更大,更优选10.0重量%或更大,特别优选20.0重量%或更大。通过icp-ms测量获得的本发明的催化剂中的贵金属的含量可以为例如60.0重量%或更小。
[0058]
当本发明的催化剂的催化剂金属颗粒各自包含贵金属合金时,通过icp-ms测量获得的本发明的催化剂中所含的贵金属与非贵金属的摩尔比(贵金属/非贵金属比)(例如,pt与非pt金属的摩尔比(pt/非pt金属比))可以为例如0.5或更大,并且优选为1.0或更大,特别优选2.0或更大。pt/非pt金属比例如可以为20.0或更小,并且优选为15.0或更小,特别优
选10.0或更小。贵金属/非贵金属比可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0059]
本发明的催化剂含有孔。具体地,本发明的催化剂在其碳载体中含有孔。本发明的催化剂的每单位重量的孔的体积(cm3/g)是通过bet法由通过使用bet法获得的氮吸附等温线获得的。另外,本发明的催化剂中的碳载体的每单位重量的孔的体积(cm3/g-载体)是通过将本发明的催化剂的每单位重量的孔的体积(cm3/g)除以通过下式计算出的本发明的催化剂中所含碳载体的重量比而计算出的:碳载体的重量比=1-(通过icp-ms获得的金属含量(重量%))/100。
[0060]
本发明的催化剂含有每个具有0.5nm或更大且2.0nm或更小的直径的第一孔。第一孔是本发明的催化剂种所含的碳载体的孔之中相对小的孔。第一孔预期用作每个具有相对小的粒径并且表现出高催化活性的催化剂金属颗粒的成核和颗粒生长的场所。
[0061]
本发明的催化剂的每单位重量的第一孔的体积可以为0.15(cm3/g)或更大,并且优选为0.20(cm3/g)或更大,特别优选0.25(cm3/g)或更大。
[0062]
本发明的催化剂的每单位重量的第一孔的体积可以为例如3.00(cm3/g)或更小、2.00(cm3/g)或更小,或1.00(cm3/g)或更小。本发明的催化剂的每单位重量的第一孔的体积可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0063]
本发明的催化剂的碳载体的每单位重量的第一孔的体积为0.20(cm3/g-载体)或更大。碳载体的每单位重量的第一孔的体积为例如优选0.25(cm3/g-载体)或更大,更优选0.30(cm3/g-载体)或更大,特别优选0.35(cm3/g-载体)或更大。
[0064]
碳载体的每单位重量的第一孔的体积可以为例如3.00(cm3/g-载体)或更小、2.00(cm3/g-载体)或更小,或1.00(cm3/g-载体)或更小。碳载体的每单位重量的第一孔的体积可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0065]
第一孔的大体积有助于本发明的催化剂的催化活性。即,例如,当本发明的催化剂用作氧还原催化剂时,第一孔的大体积提供了优先生成各自表现出高催化活性且具有相对小粒径的催化剂金属颗粒的场所,并且因此有助于本发明的催化剂的优异氧还原催化活性。
[0066]
本发明的催化剂含有每个具有大于2.0nm且4.0nm或更小的直径的第二孔。第二孔是相对小但比第一孔大的孔。第二孔预期用作输送反应物和本发明催化剂的化学反应产物的场所。
[0067]
本发明的催化剂的每单位重量的第二孔的体积可以为0.15(cm3/g)或更大,并且特别优选0.20(cm3/g)或更大。本发明的催化剂的每单位重量的第二孔的体积可以为例如3.00(cm3/g)或更小、2.50(cm3/g)或更小、2.00(cm3/g)或更小、1.50(cm3/g)或更小,或1.00(cm3/g)或更小。本发明的催化剂的每单位重量的第二孔的体积可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0068]
本发明的催化剂的碳载体的每单位重量的第二孔的体积为0.20(cm3/g-载体)或更大。碳载体的每单位重量的第二孔的体积优选为0.25(cm3/g-载体)或更大,更优选0.30(cm3/g-载体)或更大,还更优选0.33(cm3/g-载体)或更大,特别优选0.35(cm3/g-载体)或更大。
[0069]
碳载体的每单位重量的第二孔的体积可以为例如3.00(cm3/g-载体)或更小、2.50
(cm3/g-载体)或更小、2.00(cm3/g-载体)或更小、1.50(cm3/g-载体)或更小,或1.00(cm3/g-载体)或更小。碳载体的每单位重量的第二孔的体积可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0070]
第二孔的大体积有助于本发明的催化剂的耐久性。即,例如,当本发明的催化剂用作燃料电池电极的氧还原催化剂时,预期第二孔促进所产生水的排出。因此,当第二孔的体积较大时,本发明的催化剂的碳载体中产生的水的滞留和伴随的碳的氧化腐蚀得到有效抑制,并且催化剂金属颗粒的聚集和脱附得以抑制。此外,预期第二孔相对牢固地负载催化剂金属颗粒,因此第二孔的大体积有效地抑制催化剂金属颗粒从本发明的催化剂的碳载体的聚集和脱附。
[0071]
本发明的催化剂可以含有每个具有大于4.0nm且50.0nm或更小的直径的第二孔。第三孔是比第二孔大的孔。第三孔提供负载每个具有相对大粒径的催化剂金属颗粒的场所。
[0072]
本发明的催化剂的每单位重量的第三孔的体积可以为例如0.20(cm3/g)或更大,并且优选0.25(cm3/g)或更大,特别优选0.30(cm3/g)或更大。本发明的催化剂的每单位重量的第三孔的体积可以为例如3.00(cm3/g)或更小、2.50(cm3/g)或更小、2.00(cm3/g)或更小、1.50(cm3/g)或更小,或1.00(cm3/g)或更小。进一步地,本发明的催化剂的每单位重量的第三孔的体积例如优选为0.75(cm3/g)或更小,更优选0.70(cm3/g)或更小,还更优选0.65(cm3/g)或更小,特别优选0.60(cm3/g)或更小。本发明的催化剂的每单位重量的第三孔的体积可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0073]
本发明的催化剂的碳载体的每单位重量的第三孔的体积可以为例如0.25(cm3/g-载体)或更大。碳载体的每单位重量的第三孔的体积例如优选为0.30(cm3/g-载体)或更大,更优选0.35(cm3/g-载体)或更大,还更优选0.40(cm3/g-载体)或更大,特别优选0.45(cm3/g-载体)或更大。
[0074]
碳载体的每单位重量的第三孔的体积可以为例如3.00(cm3/g-载体)或更小、2.50(cm3/g-载体)或更小、2.00(cm3/g-载体)或更小、1.50(cm3/g-载体)或更小,或1.00(cm3/g-载体)或更小。进一步地,碳载体的每单位重量的第三孔的体积为例如优选0.90(cm3/g-载体)或更小,更优选0.80(cm3/g-载体)或更小,特别优选0.70(cm3/g-载体)或更小。碳载体的每单位重量的第三孔的体积可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0075]
本发明的催化剂的每单位重量的第三孔的体积与第二孔的体积的比率(第三孔/第二孔体积比)可以为例如3.00或更小,并且优选2.50或更小,特别优选2.00或更小。
[0076]
本发明的催化剂的每单位重量的第三孔/第二孔体积比可以为例如0.1或更大,或0.5或更大。本发明的催化剂的每单位重量的第三孔/第二孔体积比可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0077]
本发明的催化剂的碳载体的每单位重量的第三孔的体积与第二孔的体积的比率(第三孔/第二孔体积比)可以为例如3.00或更小。本发明的催化剂的碳载体的每单位重量的第三孔/第二孔体积比可以为例如优选2.50或更小,特别优选2.00或更小。
[0078]
第三孔/第二孔体积比可以为例如0.1或更大,或0.5或更大。本发明的催化剂的第三孔/第二孔体积比可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合
来指定。
[0079]
第三孔/第二孔体积比不太大时,每个具有大粒径的催化剂金属颗粒的比例的过度增加被有效地抑制。
[0080]
本发明的催化剂的通过xps测量的贵金属(例如pt)的含量(重量%)与通过icp-ms测量的贵金属的含量(重量%)的比率(贵金属-xps/icp比)为0.35或更大且0.75或更小。贵金属-xps/icp比是通过将通过xps获得的本发明的催化剂中的贵金属的含量(重量%)除以通过icp-ms获得的本发明的催化剂中的贵金属的含量(重量%)计算出的。
[0081]
本发明的催化剂的贵金属-xps/icp比例如优选为0.36或更大,更优选0.38或更大,特别优选0.41或更大。
[0082]
本发明的催化剂的贵金属-xps/icp比例如优选为0.70或更小,更优选0.65或更小,特别优选0.60或更小。本发明的催化剂的贵金属-xps/icp比可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0083]
在通过xps的金属测量中,除了负载在本发明的催化剂的表面(确切地说,碳载体的表面)上的金属外,还检测到距离表面(表面附近)约3nm深度的内部中负载的金属。同时,在通过icp-ms的金属测量中,检测到包含在本发明催化剂的表面上和内部处的所有金属。
[0084]
由于上述原因,小的贵金属-xps/icp比意味着,在负载在金属负载的催化剂上的金属中,包含在碳载体的较深内部(例如,距外表面3nm或更大深度处的内部)的金属的比例较大。因此,当贵金属-xps/icp比太小时,对催化活性贡献不足的金属的比例增加。当贵金属与非贵金属形成合金时,认为贵金属和非贵金属在与本发明的催化剂中的催化剂金属相同的位置上起作用。因此,可以说本发明的催化剂的贵金属-xps/icp比表示催化剂金属的位置信息。
[0085]
同时,大的贵金属-xps/icp比意味着,在负载在金属负载的催化剂上的催化剂金属中,包含在碳载体的表面附近(表面和距表面3nm或更小的深度处的内部)的催化剂金属的比例较大。因此,当贵金属-xps/icp比太大时,容易发生由外部环境引起的催化剂金属颗粒的中毒和劣化(例如,分解和聚集)。
[0086]
在这方面,本发明的催化剂具有如上所述的特定范围内的贵金属-xps/icp比。本发明的催化剂的贵金属-xps/icp比落在上述特定范围内是指,在负载在本发明的催化剂上的催化剂金属颗粒中,特定适当比例的催化剂金属颗粒包含在碳载体的表面附近(表面和距表面3nm或更小的深度处的内部),并且以剩余特定适当比例的催化剂金属颗粒包含在碳载体的较深内部(例如距外表面3nm或更大的深度处的内部)。即,具有在上述特定范围内的贵金属-xps/icp比的本发明的催化剂含有对催化活性有充分贡献并且不易被外部环境中毒或劣化的催化剂金属颗粒。
[0087]
在本发明的催化剂中,使用通过分离在由cukα射线得到的粉末x射线衍射图中衍射角2θ在40
°
附近的衍射线而获得的一个或多个衍射峰的衍射角和半峰全宽由谢乐方程计算出的所述催化剂金属颗粒的平均微晶直径可以为例如7.0nm或更小。
[0088]
通过xrd测量的催化剂金属颗粒的平均微晶直径例如优选为6.5nm或更小,更优选6.0nm或更小,并且特别优选5.5nm或更小。此外,催化剂金属颗粒的平均微晶直径可以为例如1.0nm或更大。
[0089]
通过xrd的催化剂金属颗粒的平均微晶直径可以通过将上述下限值和上述上限值
中的任一个任意组合来指定。催化剂金属颗粒的平均微晶直径通过后述的实施例中描述的方法确定。
[0090]
当负载在碳载体上的催化剂金属颗粒的粒径较小时,催化剂金属颗粒的比表面积增加。因此,大比例的每个具有较小粒径的催化剂金属颗粒有助于本发明催化剂的催化活性。
[0091]
在本发明的催化剂中,其中平均微晶直径为5.0nm或更小的催化剂金属颗粒的量相对于催化剂金属颗粒的总量的比例可以为65%或更大,该平均微晶直径是通过谢乐方程,通过使用在通过cukα射线得到的粉末x射线衍射图中通过分离具有在40
°
附近的衍射角2θ的衍射线而获得的一个或多个衍射峰(例如,碳载体的衍射峰、纯贵金属颗粒的衍射峰,和贵金属合金颗粒的一个或多个衍射峰)的衍射角和半峰全宽而计算的。
[0092]
通过xrd测量的平均微晶直径为5.0nm或更小的催化剂金属颗粒的比例例如优选为70%或更大,更优选75%或更大,特别优选80%或更大。
[0093]
每个具有5.0nm或更小的通过用透射电子显微镜(tem)观察本发明的催化剂获得的粒径的催化剂金属颗粒的比例(%)可以为,例如,60%或更大,并且优选为65%或更大,更优选70%或更大,还更优选75%或更大,并且特别优选80%或更大。
[0094]
每个具有5.0nm或更小的通过tem观察获得的粒径的催化剂金属颗粒的比例(%)是通过将一个值乘以100而计算出的,该值是通过在本发明的催化剂的tem图像中随机选择的100个催化剂金属颗粒中,将每个具有5.0nm或更小的其最长部分的长度的催化剂颗粒催化剂颗粒的数量除以100(其是催化剂颗粒的总数量)获得的。
[0095]
在本发明的催化剂的催化剂金属颗粒中,大比例的每个具有5.0nm或更小的粒径的催化剂金属颗粒有助于优异的催化活性。每个具有5.0nm或更小的粒径的催化剂金属颗粒具有大的粗糙度系数,并且因此物质传输阻力降低。例如,当本发明的催化剂用作燃料电池的氧还原催化剂时,氧传输阻力降低。结果,降低了电压损失,并获得了更大的最大输出。
[0096]
本发明的催化剂的平均粒径可以为例如1.00μm或更小,并且优选0.80μm或更小,更优选0.60μm或更小,还更优选0.40μm或更小,特别优选0.30μm或更小。本发明的催化剂的平均粒径可以为例如0.01μm或更大。本发明的催化剂的平均粒径通过激光衍射法测定。
[0097]
本发明的催化剂的平均粒径等于或小于上述特定阈值有助于通过本发明的催化剂增加化学反应效率,有助于本发明的催化剂的优异催化活性,并且还有助于增加含有本发明的催化剂的电池电极的生产效率。
[0098]
金属负载的催化剂的stem二次电子图像中催化剂金属颗粒的数量与金属负载的催化剂的haadf-stem图像中催化剂金属颗粒的数量的比例(二次电子图像/haadf图像比例(%))可以为例如12%或更小,优选为11%或更小,特别优选10%或更小。
[0099]
二次电子图像/haadf图像比例(%)等于或小于上述特定阈值有助于本发明的催化剂的催化活性。即,例如,在将本发明的催化剂用作燃料电池电极的氧还原催化剂的情况下,在二次电子图像/haadf图像比例(%)等于或小于上述特定阈值时,存在于碳载体的最外表面上的催化剂金属的比例降低时,因此抑制了电解质等引起的催化剂金属的中毒。
[0100]
本发明的催化剂的bet比表面积可以为例如400m2/g或更大,优选500m2/g或更大,更优选600m2/g或更大,特别优选800m2/g或更大。
[0101]
本发明的催化剂的bet比表面积可以为例如3.000(m2/g)或更小,或2,500m2/g或更
小。本发明的催化剂的bet比表面积可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0102]
通过将本发明的催化剂的bet比表面积乘以本发明的催化剂的重量与本发明的催化剂中所含的碳载体的重量之比而计算出的归一化比表面积可以为例如1,100(m2/g-载体)或更大。
[0103]
本发明的催化剂的归一化比表面积例如优选为1,150(m2/g-载体)或更大,更优选1,200(m2/g-载体)或更大,还更优选1,250(m2/g-载体)或更大,特别优选1,300(m2/g-载体)或更大。
[0104]
本发明的催化剂的归一化比表面积可以为例如3,000(m2/g-载体)或更小,或2,500(m2/g-载体)或更小。本发明的催化剂的归一化比表面积可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0105]
本发明的催化剂的大比表面积(bet比表面积和/或归一化比表面积,特别是归一化比表面积)有助于其催化活性。当比表面积增加时,催化剂金属颗粒可能更均匀地负载在碳载体内侧上的孔中。因为如此,例如,当本发明的具有大比表面积的催化剂用作燃料电池的电极催化剂时,催化剂金属颗粒的聚集和催化剂金属颗粒被电解质覆盖得以有效抑制。当电解质的覆盖被抑制时,可以有效地利用催化剂金属颗粒。
[0106]
在本发明的催化剂中,通过xps测得的氮原子浓度(原子%)与碳原子浓度(原子%)的比率(xps-n/c比)可以为,例如,0.010或更大。通过xps测得的n/c比为例如优选0.011或更大,特别优选0.012或更大。本发明的催化剂的通过xps测得的n/c比可以为例如0.15或更小。另外,氮原子含量(重量%)与碳原子含量(重量%)的比率(xps-n/c重量比)由碳原子和氮原子的原子量比确定。
[0107]
在本发明的催化剂中,通过使用燃烧法的元素分析(chn)测得的氮原子含量(重量%)与碳原子含量(重量%)的比率(chn—n/c比)可以为0.010或更大,优选0.015或更大,特别优选0.020或更大。本发明的催化剂的chn-n/c比可以为例如0.20或更小。
[0108]
等于或大于上述特定阈值的本发明的催化剂的n/c比反映了本发明的催化剂的碳结构(更具体地,碳载体的碳结构)的表面含有大量的非催化剂金属活性位点(碳载体本身的活性位点,而不是负载在碳载体上的催化剂金属活性位点),诸如含氮官能团、特定类型的氮原子(诸如吡啶型氮、吡咯型氮、和引入碳网络平面内部的石墨型氮),以及由上述形成的碳弯曲结构,并且有助于本发明的催化剂的优异催化活性。
[0109]
本发明的催化剂的chn-n/c比与xps-n/c重量比的比率(n-chn/xps比)可以为例如0.90或更大。n-chn/xps比例如优选为1.00或更大,更优选1.10或更大,特别优选1.20或更大。n-chn/xps比可以为例如3.00或更小。
[0110]
本发明催化剂的高n-chn/xps比有助于本发明的催化剂的耐久性。通过xps测得的氮原子含量表示存在于碳载体的表面上且在距表面数纳米深度的范围内的氮原子的含量。同时,通过使用燃烧法的元素分析测得的氮原子含量表示包括碳载体的表面和内部在内的整体中存在的氮原子的含量。
[0111]
因此,本发明的催化剂的高n-chn/xps比表明,与碳载体表面相比,碳载体内部存在大量氮原子。氮原子与金属结合。因此,当氮原子存在于碳载体内部时,催化剂金属颗粒负载在碳载体内部,并且抑制了催化剂金属的溶出。结果,提高了本发明的催化剂的耐久
性。
[0112]
本发明的催化剂可具有碳结构,该碳结构在通过拉曼光谱法获得的拉曼光谱中表现出,在1,360cm-1
附近(例如在1,250cm-1
或更大且1,450cm-1
或更小的范围内)具有峰顶的d带的160cm-1
或更小的半峰全宽。
[0113]
即,在这种情况下,在本发明的催化剂的通过拉曼光谱法获得的拉曼光谱中,在1,360cm-1
附近(例如,1,250cm-1
或更大且1,450cm-1
或更小)检测到半峰全宽为160cm-1
或更小的d带。
[0114]
另外,在这种情况下,优选的是,本发明的催化剂的通过xps测得的n/c比等于或大于上述下限值,和/或本发明的催化剂的通过使用燃烧法的元素分析测得的n/c比率等于或大于上述下限值。
[0115]
本发明的催化剂的d带的半峰全宽例如优选为155cm-1
或更小,特别优选150cm-1
或更小。本发明的催化剂的d带的半峰全宽可以例如为80cm-1
或更大。
[0116]
d带的半峰全宽由下式计算:d带半峰全宽(cm-1
)=(a
d-bd)
×
2(在该等式中,ad表示与d带强度id(d带的峰顶)相对应的拉曼位移(cm-1
),并且bd表示与在比ad较低的波数侧上表现出d带强度id的一半的强度的拉曼光谱相对应的拉曼位移(cm-1
)。
[0117]
在拉曼光谱中,d带的半峰全宽表示包含在碳结构中的弯曲结构的结晶度。即,d带的小的半峰全宽意味着弯曲结构的结晶度高。因为如此,本发明的催化剂的碳结构(具体地,碳载体的碳结构)的d带的半峰全宽等于或小于上述特定阈值意味着碳结构包含高度结晶的弯曲结构。具有包含高度结晶的弯曲结构的碳结构的本发明的催化剂有助于本发明的催化剂的优异耐久性和抗氧化性。
[0118]
本发明的催化剂可具有碳结构,该碳结构表现出在通过拉曼光谱法获得的拉曼光谱中,在1,600cm-1
附近具有峰顶的g带的90cm-1
或更小的半峰全宽。
[0119]
即,在这种情况下,在本发明的催化剂的通过拉曼光谱法获得的拉曼光谱中,检测到在1,600cm-1
附近(例如1,550cm-1
或更大且1,700cm-1
或更小)具有峰顶的具有90cm-1
或更小的v的g带。
[0120]
另外,在这种情况下,优选的是,本发明的催化剂的通过xps测得的n/c比等于或大于上述下限值,和/或本发明的催化剂的通过使用燃烧法的元素分析测得的n/c比等于或大于上述下限值。
[0121]
本发明的催化剂的g带的半峰全宽例如优选为85cm-1
或更小,特别优选80cm-1
或更小。本发明的催化剂的g带的半峰全宽可以例如为40cm-1
或更大。
[0122]
g带的半峰全宽由下式计算:g带半峰全宽(cm-1
)={(a
g-bg)}
×
2的绝对值(在该等式中,ag表示与g带强度ig(g带的峰顶)相对应的拉曼位移(cm-1
),并且bg表示与在ag的高波数侧上表现出g带强度ig的一半的强度的拉曼光谱相对应的拉曼位移(cm-1
)。
[0123]
在拉曼光谱中,g带的半峰全宽表示包含在碳结构中的石墨结构的结晶度。即,g带的小的半峰全宽意味着石墨结构的结晶度高。因为如此,本发明的催化剂的碳结构(具体地,碳载体的碳结构)的g带的半峰全宽等于或小于上述特定阈值意味着碳结构包含高度结晶的石墨结构。具有包含高度结晶的石墨结构的碳结构的本发明的催化剂有助于本发明的催化剂的优异耐久性和抗氧化性。
[0124]
本发明的催化剂可具有碳结构,该碳结构表现出0.20或更大且0.50或更小的在通
过拉曼光谱法获得的拉曼光谱中,在1,600cm-1
附近具有峰顶的g带和在1,360cm-1
附近具有峰顶的d带之间的最小强度与g带的强度的比率(iv/ig比)。
[0125]
即,在这种情况下,在本发明的催化剂的通过拉曼光谱法获得的拉曼光谱中,在1,360cm-1
附近检测到d带,在1,600cm-1
附近检测到g带,并且g带和d带之间的最小强度与g带的强度的比率为0.20或更大且0.50或更小。
[0126]
另外,在这种情况下,优选的是,本发明的催化剂的通过xps测得的n/c比等于或大于上述下限值,和/或本发明的催化剂的通过使用燃烧法的元素分析测得的n/c比等于或大于上述下限值。
[0127]
本发明的催化剂的iv/ig比例如优选为0.20或更大且0.45或更小,更优选0.25或更大且0.45或更小,特别优选0.25或更大且0.40或更小。
[0128]
在拉曼光谱中,g带是源自理想石墨结构的分量,d带是源自包括缺陷和边缘的弯曲结构的分量。g带和d带之间的最小强度iv取决于源自无定形物质的分量。因此,iv/ig比是无定形物质的量与理想石墨结构的量的比率。在碳结构中,活性位点存在于无定形物质中。然而,当无定形物质的量太大时,碳载体(例如作为碳催化剂的碳载体)容易劣化,并且因此认为存在iv/ig比的最佳范围。在这方面,本发明的催化剂的碳结构(具体地,碳载体的碳结构)的iv/ig比落入上述特定范围内意味着碳载体具有除了本发明的催化剂中的金属之外的优异的催化剂活性位点(非催化剂金属活性位点)。
[0129]
本发明的催化剂可以具有碳结构,该碳结构在程序升温脱附法(tpd)中表现出0.85
×
10-5
(mol/g)或更大的本发明的催化剂的每单位重量的从600℃到1,000℃的氮脱附量。另外,本发明的催化剂可以具有例如碳结构,该碳结构在tpd中表现出1.00
×
10-1
(mol/g)或更小的本发明的催化剂的每单位重量的从600℃到1,000℃的氮脱附量。
[0130]
另外,在这种情况下,优选的是,本发明的催化剂的通过xps测得的n/c比等于或大于上述下限值,和/或本发明的催化剂的通过使用燃烧法的元素分析测得的n/c比等于或大于上述下限值。
[0131]
在本发明的催化剂的tpd中,本发明的催化剂的每单位重量的从600℃到1,000℃的氮脱附量例如优选为3.00
×
10-5
(mol/g)或更大,更优选1.00
×
10-4
(mol/g)或更大,特别优选5.00
×
10-4
(mol/g)或更大。
[0132]
本发明的催化剂可以具有碳结构,该碳结构在tpd中表现出0.61
×
10-5
(mol/g)或更大的本发明的催化剂的每单位重量的从800℃到1,000℃的氮脱附量。另外,本发明的催化剂可以具有例如碳结构,该碳结构在tpd中表现出1.00
×
10-1
(mol/g)或更小的本发明的催化剂的每单位重量的从800℃到1,000℃的氮脱附量。
[0133]
另外,在这种情况下,优选的是,本发明的催化剂的通过xps测得的n/c比等于或大于上述下限值,和/或本发明的催化剂的通过使用燃烧法的元素分析测得的n/c比等于或大于上述下限值。
[0134]
在本发明的催化剂的tpd中,本发明的催化剂的每单位重量的从800℃到1,000℃的氮脱附量例如优选为2.50
×
10-5
(mol/g)或更大,更优选5.00
×
10-5
(mol/g)或更大,特别优选1.00
×
10-4
(mol/g)或更大。
[0135]
本发明的催化剂可以具有碳结构,该碳结构在tpd中表现出1.20
×
10-5
(mol/g-载体)或更大的碳载体的每单位重量的从600℃到1,000℃的氮脱附量。另外,本发明的催化剂
可以具有例如碳结构,该碳结构在tpd中表现出1.00
×
10-1
(mol/g-载体)或更小的碳载体的每单位重量的从600℃到1,000℃的氮脱附量。
[0136]
另外,在这种情况下,优选的是,本发明的催化剂的通过xps测得的n/c比等于或大于上述下限值,和/或本发明的催化剂的通过使用燃烧法的元素分析测得的n/c比等于或大于上述下限值。
[0137]
在本发明催化剂的tpd中,碳载体的每单位重量的从600℃到1,000℃的氮脱附量例如优选为5.00
×
10-5
(mol/g-载体)或更大,更优选1.00
×
10-4
(mol/g-载体)或更大,还更优选5.00
×
10-4
(mol/g-载体)或更大,特别优选1.00
×
10-3
(mol/g-载体)或更大。
[0138]
本发明的催化剂可以具有碳结构,该碳结构在tpd中表现出0.75
×
10-5
(mol/g-载体)或更大的碳载体的每单位重量的从800℃到1,000℃的氮脱附量。另外,本发明的催化剂可以具有例如碳结构,该碳结构在tpd中表现出1.00
×
10-1
(mol/g-载体)或更小的碳载体的每单位重量的从800℃到1,000℃的氮脱附量。
[0139]
另外,在这种情况下,优选的是,本发明的催化剂的通过xps测得的n/c比等于或大于上述下限值,和/或本发明的催化剂的通过使用燃烧法的元素分析测得的n/c比等于或大于上述下限值。
[0140]
在本发明催化剂的tpd中,碳载体的每单位重量的从800℃到1,000℃的氮脱附量例如优选为3.00
×
10-5
(mol/g-载体)或更大,更优选5.00
×
10-5
(mol/g-载体)或更大,特别优选1.00
×
10-4
(mol/g-载体)或更大。
[0141]
被定义为本发明的催化剂的特征之一的tpd中的氮脱附量反映了本发明的催化剂的碳结构(具体地,碳载体的碳结构)中所含的氮原子的质量和量。即,本发明的催化剂通过具有包含特定质量和量的氮原子的碳结构以使得在上述相对高的特定温度范围内的氮脱附量在上述tpd中表现出上述特定阈值或更低而表现出优异的耐久性。
[0142]
以下特征是指本发明的催化剂的碳载体具有除催化剂金属以外的活性位点(非催化剂金属活性位点)并且具有优异的抗氧化性:本发明的催化剂的n/c比(具体地,碳载体的xps-n/c比或chn-n/c比)具有上述特定的下限值或更大;在本发明的催化剂的通过拉曼光谱法获得的拉曼光谱中,d带的半峰全宽具有上述特定的上限值或更小,g带的半峰全宽具有上述特定的上限值或更小,或iv/ig比落在上述特定范围内;并且,在本发明的催化剂的tpd中,600℃到1,000℃的氮脱附量具有上述特定的下限值或更大,或800℃到1,000℃的氮脱附量具有上述特定的下限值或更大。
[0143]
在碳载体(例如作为碳催化剂的碳载体)具有非催化剂金属活性位点的情况下,例如,当本发明的催化剂用作燃料电池电极的氧还原催化剂时,在高负载操作期间,氧在催化剂金属上的浓度降低,并且高电流密度区域中的电压降低得到抑制。另外,在碳载体具有优异的抗氧化性的情况下,当由于操作期间的负载波动、启动-停止等而施加高电位时碳载体的氧化腐蚀得到抑制。结果,减轻了催化剂金属(例如,pt)的脱附和聚集,并获得了优异的耐久性。
[0144]
当本发明的催化剂的催化剂金属颗粒包括pt颗粒时,通过将在使用包含本发明的催化剂的旋转圆盘电极的循环伏安法中测量的氢吸附电量除以相对于铂的氢吸附的理论面积等效电量和负载在本发明的催化剂上的铂的重量而获得的电化学表面积(有效铂催化剂表面积:h
2-ecsa,由通过循环伏安法获得的氢吸附电量确定)可以为20.0m2/g-铂或更大,
并且通过将在使用包含本发明的催化剂的旋转圆盘电极的溶出伏安法中测量的一氧化碳吸附电量除以相对于铂的一氧化碳吸附的理论面积等效电量和负载在本发明的催化剂上的铂的重量而获得的电化学表面积(有效铂催化剂表面积:co-ecsa,由通过溶出伏安法获得的一氧化碳吸附电量确定)可以为20.0m2/g-铂或更大。
[0145]
本发明的催化剂表现出例如优选25.0m2/g-铂或更大的h
2-ecsa和30.0m2/g-铂或更大的co-ecsa,更优选30.0m2/g-铂或更大的h
2-ecsa和40.0m2/g-铂或更大的co-ecsa,特别优选35.0m2/g-铂或更大的h
2-ecsa和50.0m2/g-铂或更大的co-ecsa。
[0146]
本发明的催化剂可以表现出例如200m2/g-铂或更小的h
2-ecsa和200m2/g-铂或更小的co-ecsa。本发明的催化剂的h
2-ecsa和co-ecsa可以通过将上述下限值中的任一个和上述上限值任意组合来指定。
[0147]
本发明的催化剂的大h
2-ecsa和co-ecsa有助于优异的催化活性。例如,在本发明的催化剂用作燃料电池电极的氧还原催化剂的情况下,当催化剂金属颗粒的h
2-ecsa和co-ecsa小时,出现氧浓度阻力,导致输出下降(kongkanand,a.;subramanian,n.p.;yu,y.;liu,z.;igarashi,h.;muller,d.a.achieving high-power pem fuel cell performance with an ultralow-pt-content core-shell catalyst.acs catal.2016,6,1578-1583.)。同时,当本发明的催化剂的h
2-ecsa和co-ecsa增加时,氧浓度阻力降低。结果,降低了电压损失,并获得了更大的最大输出。
[0148]
本发明的催化剂的碳载体中的非催化剂金属活性位点的铂当量可以为例如1.0(mg-pt/g-载体)或更大,并且优选为3.0(mg-pt/g-载体)或更大,特别优选5.0(mg-pt/g-载体)或更大。
[0149]
本发明的催化剂的大量非催化剂金属活性位点有助于优异的催化活性。例如,当用作燃料电池电极的氧还原催化剂的本发明的催化剂的活性位点仅为催化剂金属颗粒时,氧过度聚集在催化剂金属颗粒上,并且本发明的催化剂的催化活性降低。当本发明的催化剂具有非催化剂金属活性位点时(具体地,当碳载体本身具有活性位点时),可以抑制催化剂金属颗粒上的过高氧浓度,并且改善本发明的催化剂的催化活性。
[0150]
本发明的催化剂是通过使碳载体负载催化剂金属颗粒来生产的。生产本发明的催化剂的方法包括用催化剂金属浸渍碳载体并通过使用电磁波来加热浸渍有催化剂金属的碳载体以形成负载在碳载体上的催化剂金属的颗粒。
[0151]
用于加热碳载体的电磁波没有特别限制,只要该电磁波使碳载体本身和/或浸渍到碳载体中的催化剂金属本身产生热量即可,并且例如,优选使用毫米波(超高频,extra high frequency)。毫米波是波长为1mm或更大且15mm或更小的电磁波。
[0152]
当形成负载在碳载体上的贵金属合金颗粒时,用贵金属(例如,pt)和与该贵金属形成合金的非贵金属(例如,过渡金属)浸渍碳载体。在这种情况下,例如,优选的是,首先用非贵金属浸渍碳载体,并且然后用贵金属浸渍浸渍有非贵金属的碳载体。
[0153]
将催化剂金属浸渍到碳载体中是通过例如将碳载体浸入含有催化剂金属的溶液中来进行的。即,当形成负载在碳载体上的贵金属合金颗粒时,将碳载体浸入含有贵金属的溶液和含有非贵金属的溶液中的每一种中。在这种情况下,优选的是,首先将碳载体浸入含有非贵金属的溶液中,然后将浸渍有非贵金属的碳载体浸入含有贵金属的溶液中。
[0154]
更具体地,首先将碳载体浸入含有非贵金属的溶液中。然后,将浸渍有非贵金属的
碳载体干燥,并进一步将浸渍有非贵金属并干燥的碳载体浸渍在含有贵金属的溶液中。之后,将浸渍有非贵金属和贵金属的碳载体干燥。
[0155]
通过使用电磁波(例如,毫米波)加热浸渍有催化剂金属的碳载体的方法没有特别限制,只要该方法涉及用电磁波照射碳载体,从而使碳载体本身和/或催化剂金属本身产生热量即可。
[0156]
在通过使用电磁波(例如,毫米波)加热碳载体中,升温速率可以为例如10℃/min或更高,并且优选为50℃/min或更高,更优选100℃/min或更高,特别优选200℃/min或更高。在通过使用毫米波加热碳载体中,升温速率可以为例如1,000℃/min或更小。
[0157]
在通过使用电磁波(例如,毫米波)加热碳载体中,可以将碳载体加热至200℃或更高的温度,并且优选加热至温度300℃或更高的温度,更优选500℃或更高的温度,特别优选700℃或更高的温度。
[0158]
在通过使用电磁波(例如,毫米波)加热碳载体中,可以将碳载体加热至1,500℃或更低的温度,并且优选加热至温度1,200℃或更低的温度,特别优选1,000℃或更低的温度。通过使用电磁波加热碳载体的温度可以通过将上述下限值中的任一个和上述上限值中的任一个任意组合来指定。
[0159]
在通过使用电磁波(例如,毫米波)加热碳载体中,可以将碳载体在上述加热温度下保持1秒或更长时间,并且优选保持10分钟或更长时间。在通过使用电磁波加热碳载体中,将碳载体保持在上述加热温度下的时间可以为例如24小时或更短。
[0160]
通过使用电磁波(例如,毫米波)加热碳载体优选在还原气氛中进行,并且优选在氢气气氛中进行。
[0161]
碳载体是通过例如碳化包含有机物质的原料来生产的。这里,将描述通过包括在加压下碳化包含有机物质的原料的方法生产作为碳催化剂的碳载体的情况。
[0162]
原料中所含的有机物质没有特别限制,只要该有机物质可以被碳化即可。即,作为有机物质,使用例如高分子量有机化合物(例如,树脂,诸如热固性树脂和/或热塑性树脂)和/或低分子量有机化合物。另外,生物质可以用作有机物质。
[0163]
作为有机物质,优选使用含氮有机物质。含氮有机物质没有特别限制,只要该有机物质包含在分子中含有氮原子的有机化合物即可。当碳催化剂是包含含氮有机物质的原料的碳化产物时,碳催化剂的碳结构包含氮原子。
[0164]
具体地,例如,选自由以下组成的组一种或多种用作有机物质:聚丙烯腈、聚丙烯腈-聚丙烯酸共聚物、聚丙烯腈-聚甲基丙烯酸酯、聚丙烯腈-聚甲基丙烯酸共聚物、聚丙烯腈-聚甲基丙烯酸-聚甲基烯丙基磺酸共聚物、聚丙烯腈-聚甲基丙烯酸甲酯共聚物、酚树脂、聚糠醇、呋喃、呋喃树脂、酚醛树脂、三聚氰胺、三聚氰胺树脂、环氧树脂、含氮螯合树脂(例如,选自由多胺型、亚氨基二乙酸型、氨基磷酸型和氨基甲基膦酸型螯合树脂组成的组的一种或多种)、聚酰胺-酰亚胺树脂、吡咯、聚吡咯、聚乙烯吡咯、3-甲基聚吡咯、丙烯腈、聚偏二氯乙烯、噻吩、恶唑、噻唑、吡唑、乙烯基吡啶、聚乙烯吡啶、哒嗪、嘧啶、哌嗪、吡喃、吗啉、咪唑、1-甲基咪唑、2-甲基咪唑、喹喔啉、苯胺、聚苯胺、琥珀酸二酰肼、己二酸二酰肼、聚砜、聚氨基双马来酰亚胺、聚酰亚胺、聚乙烯醇、聚乙烯醇缩丁醛、苯并咪唑、聚苯并咪唑、聚酰胺、聚酯、聚乳酸、聚醚、聚醚醚酮、纤维素、羧甲基纤维素、木质素、几丁质、壳聚糖、沥青、褐煤、丝绸、羊毛、聚氨基酸、核酸、dna、rna、肼、酰肼、尿素、salen、聚咔唑、聚双马来酰亚
胺、三嗪、聚丙烯酸、聚丙烯酸酯、聚甲基丙烯酸酯、聚甲基丙烯酸、聚氨酯、聚酰胺胺和聚碳二亚胺。
[0165]
原料中有机物质的含量没有特别限制,只要该含量落在获得碳催化剂的范围内即可,但可以例如为5质量%或更大且90质量%或更小,并且优选为10质量%或更大且80质量%或更小。
[0166]
用于碳化的原料可以进一步包含金属。即,在这种情况下,包含有机物质和金属的原料在加压下被碳化。当碳催化剂是通过将包含有机物质和金属的原料碳化而获得的碳化材料时,碳催化剂包含金属。
[0167]
原料中所含的金属(即碳催化剂中所含的金属)优选为过渡金属。原料可以含有一种过渡金属,或者可以含有两种或更多种过渡金属。
[0168]
在本实施方案中,过渡金属可以是元素周期表中第iii-xii族的过渡金属,优选是元素周期表中第iii-xii族第四周期的过渡金属。具体地,原料中所含的过渡金属可以是例如选自由ti、v、cr、mn、fe、co、ni、cu、zn、y、zr、nb、mo、ru、rh、pd、ag、镧系元素(例如gd)和锕系元素组成的组,或者由sc、ti、v、cr、mn、fe、co、ni、cu、zn、y、zr、nb、mo、ag、镧系元素(例如gd)和锕系元素组成的组的一种或更多种或两种或更多种。
[0169]
另外,过渡金属优选是选自由fe、co、ni、cu和zn组成的组的一种或更多种或者两种或更多种,特别优选选自由fe、co、ni和zn组成的组的一种或更多种或两种或更多种。
[0170]
原料可以不包含pt。在这种情况下,原料可以不包含选自由pt、ru、rh、pd、ir、au和os组成的组的一种或更多种。
[0171]
作为原料中所含的金属,使用金属的单质和/或金属的化合物。作为金属化合物,可以使用例如选自由金属盐、金属氧化物、金属氢氧化物、金属氮化物、金属硫化物、金属碳化物和金属络合物组成的组的一种或多种。
[0172]
原料中金属的含量(当使用两种或更多种金属时,两种或更多种金属的含量之和)没有特别限制,只要该含量落在获得本发明的催化剂的范围内即可,但可以例如为1质量%或更大且90质量%或更小,并且优选为2质量%或更大且80质量%或更小。
[0173]
通过加热原料并将原料保持在原料被碳化的温度(下文中称为“碳化温度”)下而在加压下进行碳化。碳化温度没有特别限制,只要原料被碳化即可。碳化温度例如为300℃或更高。即,在这种情况下,包含有机物质的原料在加压下在300℃或更高的温度下被碳化。
[0174]
另外,碳化温度可以为例如700℃或更高,并且优选为900℃或更高,更优选1,000℃或更高,特别优选1,100℃或更高。碳化温度的上限值没有特别限制,但碳化温度例如为3,000℃或更低。
[0175]
升高至碳化温度的升温速率例如为0.5℃/分钟或更大且300℃/分钟或更小。在碳化中,不一定需要将原料保持在碳化温度下,但将原料保持在碳化温度下的时间可以为例如1秒或更长且24小时或更短,或5分钟或更长且24小时或更短。碳化优选在惰性气体气氛诸如氮气气氛中进行。即,优选在惰性气体诸如氮气的循环下进行碳化。
[0176]
碳化气氛的压力没有特别限制,只要该压力大于大气压力即可,并且例如,在表压方面为0.05mpa或更高的压力。另外,碳化气氛的压力在表压方面可以为0.15mpa或更高,并且优选为0.20mpa或更高,更优选0.40mpa或更高,特别优选0.50mpa或更高。即,在这些情况下,在碳催化剂的生产中,包含有机物质的原料在表压等于或大于上述阈值(mpa)的加压下
被碳化。
[0177]
一种生产碳催化剂的方法可以包括对通过碳化包含有机物质的原料而获得的碳化材料进行进一步处理。即,例如,可以对碳化材料进行氨处理。在这种情况下,例如,将包含有机物质的原料在加压下碳化,并对通过碳化获得的碳化材料进行氨处理。
[0178]
氨处理没有特别限制,只要该氨处理涉及使碳化材料与氨接触即可。即,氨处理是例如在含氨的气体气氛中加热碳化材料的处理。
[0179]
含氨气体的氨含量没有特别限制,只要获得该氨处理的效果即可,但可以为例如0.1体积%或更高、1.0体积%或更高,或3.0体积%或更高。
[0180]
氨处理期间加热碳化材料的温度没有特别限制,只要获得该氨处理的效果即可,但可以例如为300℃或更高,并且优选为500℃或更高,特别优选700℃或更高。加热温度的上限值没有特别限制,但加热温度可以为例如1300℃或更低,并且优选为1000℃或更低。氨处理期间加热温度的范围通过将上述下限值中的每一个和上述上限值中的每一个任意组合来定义。
[0181]
另外,可以对碳化材料进行金属去除处理。在这种情况下,例如,将包含有机物质的原料在加压下碳化,然后对通过碳化获得的碳化材料进行金属去除处理。另外,例如,将包含有机物质的原料在加压下碳化。然后,对通过碳化获得的碳化材料进行金属去除处理。之后,对金属去除处理后的碳化材料进行氨处理。金属去除处理是用于减少碳化材料中所含的源自原料的金属的量的处理。金属去除处理例如是酸洗处理和/或电解处理。
[0182]
本发明的电极包括本发明的上述催化剂。即,本发明的电极是例如承载本发明的催化剂的电池电极。具体地,本发明的电极例如是包括电极基材和负载在电极基材上的本发明的催化剂的电池电极。
[0183]
本发明的电极例如是用于燃料电池(例如,聚合物电解质燃料电池)、空气电池、水电解槽(例如,聚合物电解质水电解槽)、氧化还原液流电池或卤素电池的电极。另外,本发明的电极例如为阴极或阳极,优选为阴极。即,本发明的电极为燃料电池、空气电池、水电解槽、氧化还原液流电池或卤素电池的阴极或阳极,优选为燃料电池阴极、空气电池阴极、水电解槽阴极、氧化还原液流电池阴极或卤素电池阴极。
[0184]
本发明的电池包括上述电池电极。即,本发明的电池例如是包括本发明的电极的燃料电池(例如,聚合物电解质燃料电池)、空气电池、氧化还原液流电池或卤素电池。本发明的电池可以包括包含本发明的电极的膜电极组件(mea)。本发明的电池是包括本发明的电极作为阴极或阳极的电池,优选包括本发明的电极作为阴极的电池。即,本发明的电池是包括本发明的电极作为阴极或阳极的燃料电池、空气电池、氧化还原液流电池或卤素电池,优选为包括本发明的电极作为阴极的燃料电池、空气电池、氧化还原液流电池或卤素电池。
[0185]
接下来,将描述根据本发明的实施方案的具体实施例。
[0186]
实施例
[0187]
[碳载体a]
[0188]
将1.0g聚丙烯腈(pan)、1.0g 2-甲基咪唑、6.0g氯化锌(zncl2)、0.18g氯化铁(iii)六水合物(fecl3·
6h2o)和30g二甲基甲酰胺混合。通过干燥从所得混合物中除去溶剂。将干燥的混合物在大气中加热以在250℃下进行不熔化。
[0189]
通过在氮气气氛中在0.90mpa的表压下将不熔化的混合物加热并保持在1,300℃
下,将不熔化的混合物碳化。将稀盐酸加入到通过碳化获得的碳化材料中,并搅拌混合物。然后,通过使用过滤膜来过滤含有碳化材料的悬浮液,并用蒸馏水洗涤碳化材料直至滤液变为中性。以此方式,通过用酸洗涤进行金属去除处理。
[0190]
将金属去除处理后的碳化材料用细粉磨机粉碎,直到其粒径中值达到300nm或更小。将硝酸加入粉碎后的碳化材料中,并搅拌混合物。然后,通过使用过滤膜来过滤含有碳化材料的悬浮液,并用蒸馏水洗涤碳化材料直至滤液变为中性。以此方式,用硝酸进行氧化处理。
[0191]
将氧化处理后的碳化材料在100%氨气以0.15l/min循环的气氛中以50℃/min的升温速率加热,并保持在900℃下1小时。之后,用氮气置换氨气,并将碳化材料在氮气气氛中保持在500℃下10分钟。以此方式,用氨气进行氨处理。然后,获得通过在氮气气氛中自然冷却而冷却的碳化材料作为碳载体a。
[0192]
[碳载体b]
[0193]
通过与上述碳载体a相同的方法获得碳载体b,不同之处在于使用12.0g氯化锌(zncl2)代替6.0g氯化锌。
[0194]
[碳载体c]
[0195]
通过与上述碳载体a相同的方法获得碳载体c,不同之处在于不进行硝酸氧化处理和氨处理。
[0196]
[碳载体d]
[0197]
将1.0g聚丙烯腈(pan)、1.0g 2-甲基咪唑、6.0g氯化锌(zncl2)、0.18g氯化铁(iii)六水合物(fecl3·
6h2o)和30g二甲基甲酰胺混合。通过干燥从所得混合物中除去溶剂。将干燥的混合物在大气中加热以在250℃下进行不熔化。
[0198]
通过在氮气气氛中在常压下将不熔化的混合物加热并保持在1,200℃下,将不熔化的混合物碳化。将稀盐酸加入到通过碳化获得的碳化材料中,并搅拌混合物。然后,通过使用过滤膜来过滤含有碳化材料的悬浮液,并用蒸馏水洗涤碳化材料直至滤液变为中性。以此方式,通过用酸洗涤进行金属去除处理。
[0199]
将金属去除处理后的碳化材料用细粉磨机粉碎,直到其粒径中值达到600nm或更小。将硝酸加入粉碎后的碳化材料中,并搅拌混合物。然后,通过使用过滤膜来过滤含有碳化材料的悬浮液,并用蒸馏水洗涤碳化材料直至滤液变为中性。以此方式,用硝酸进行氧化处理。
[0200]
将氧化处理后的碳化材料在100%氨气以0.15l/min循环的气氛中以50℃/min的升温速率加热,并保持在900℃下1小时。之后,用氮气置换氨气,并将碳化材料在氮气气氛中保持在500℃下10分钟。以此方式,用氨气进行氨处理。然后,获得通过在氮气气氛中自然冷却而冷却的碳化材料作为碳载体d。
[0201]
[碳载体e]
[0202]
通过与上述碳载体a相同的方法获得碳载体e,不同之处在于不使用氯化铁(iii)六水合物(fecl3·
6h2o)。
[0203]
[碳载体f]
[0204]
通过与上述碳载体a相同的方法获得碳载体f,不同之处在于使用0.16g氯化镍(ii)六水合物(nicl2·
6h2o)代替氯化铁(iii)六水合物(fecl3·
6h2o)。
[0205]
[碳载体g]
[0206]
通过与上述碳载体a相同的方法获得碳载体g,不同之处在于使用0.16g氯化钴(ii)六水合物(cocl2·
6h2o)代替氯化铁(iii)六水合物(fecl3·
6h2o)。
[0207]
[碳载体m]
[0208]
将硝酸加入到市售的中孔碳中,并搅拌混合物。然后,通过使用过滤膜来过滤含有中孔碳的悬浮液,并用蒸馏水洗涤中孔碳直至滤液变为中性。以此方式,用硝酸进行氧化处理。
[0209]
将氧化处理后的中孔碳在100%氨气以0.15l/min循环的气氛中以50℃/min的升温速率加热,并保持在900℃下1小时。之后,用氮气置换氨气,并将中孔碳在氮气气氛中保持在500℃下10分钟。以此方式,用氨气进行氨处理。然后,获得通过在氮气气氛中自然冷却而冷却的中孔碳作为碳载体m。
[0210]
[碳载体kb]
[0211]
通过与上述碳载体m相同的方法获得碳载体kb,不同之处在于使用科琴黑(ketjen black)ec600jd(由lion specialty chemicals co.,ltd制造)代替中孔碳。
[0212]
[碳载体v]
[0213]
通过与上述碳载体m相同的方法获得碳载体vb,不同之处在于使用vulcan xc72r(由cabot corporation制造)代替中孔碳。
[0214]
[碳载体db]
[0215]
通过与上述碳载体m相同的方法获得碳载体db,不同之处在于使用denka black hs-100(由denka company limited制造)代替中孔碳。
[0216]
[实施例1]
[0217]
通过将氯化铁(iii)六水合物(fecl3·
6h2o)溶解在水中使得铁的浓度变为0.1重量%来制备氯化铁水溶液。将碳载体a加入到氯化铁水溶液中,并将混合物搅拌16小时以获得悬浮液。过滤所获得的悬浮液,然后在100℃下干燥16小时,以获得浸渍有铁的碳载体a的粉末。
[0218]
通过将氯铂酸(h2ptcl6)溶解在水中使得铂的浓度变为1.6重量%来制备氯铂酸水溶液。将浸渍有铁的碳载体a的粉末加入到氯铂酸水溶液中,并将混合物搅拌16小时以获得悬浮液。在这种情况下,调节氯铂酸水溶液的量使得最终的铂负载量变为20重量%。过滤所获得的悬浮液,然后在100℃下干燥16小时,以获得浸渍有铂和铁的碳载体a的粉末。
[0219]
在其中氢气以100ml/min的流速循环的氢气气氛中,通过使用毫米波(辐射源:回旋振荡管,频率:28g hz,输出:1kw),将如此获得的浸渍有铂和铁的碳载体a的粉末以450℃/min的升温速率从室温加热至900℃,并保持1小时以获得金属负载的催化剂。
[0220]
通过icp-ms测量获得的金属负载的催化剂的铂负载量(负载在金属负载的催化剂上的铂的重量相对于金属负载的催化剂的重量的比例)为20重量%。另外,通过icp-ms测量获得的金属负载的催化剂中所含的铂与铁的摩尔比(pt/fe比)为5.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0221]
[实施例2]
[0222]
通过与上述实施例1相同的方法获得金属负载的催化剂,不同之处在于使用碳载
体b代替碳载体a,并且调节氯铂酸水溶液的量使得最终的铂负载量变为30重量%。通过icp-ms测量获得的金属负载的催化剂的铂负载量为20重量%。另外,通过icp-ms测量获得的金属负载的催化剂中所含的铂与铁的摩尔比(pt/fe比)为7.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0223]
[实施例3]
[0224]
通过与上述实施例1相同的方法获得金属负载的催化剂,不同之处在于使用碳载体c代替碳载体a。通过icp-ms测量获得的金属负载的催化剂的铂负载量为20重量%。另外,通过icp-ms测量获得的金属负载的催化剂中所含的铂与铁的摩尔比(pt/fe比)为5.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0225]
[实施例4]
[0226]
通过将氯化钴(ii)六水合物(cocl2·
6h2o)溶解在水中使得钴的浓度变为0.1重量%来制备氯化钴水溶液。将碳载体a加入到氯化钴水溶液中,并将混合物搅拌16小时以获得悬浮液。过滤所获得的悬浮液,然后在100℃下干燥16小时,以获得浸渍有钴的碳载体a的粉末。
[0227]
通过将氯铂酸(h2ptcl6)溶解在水中使得铂的浓度变为1.6重量%来制备氯铂酸水溶液。将浸渍有钴的碳载体a的粉末加入到氯铂酸水溶液中,并将混合物搅拌16小时以获得悬浮液。在这种情况下,调节氯铂酸水溶液的量使得最终的铂负载量变为30重量%。过滤所获得的悬浮液,然后在100℃下干燥16小时,以获得浸渍有铂和钴的碳载体a的粉末。
[0228]
在其中氢气以100ml/min的流速循环的氢气气氛中,通过使用毫米波(辐射源:回旋振荡管,频率:28g hz,输出:1kw),将如此获得的浸渍有铂和钴的碳载体a的粉末以450℃/min的升温速率从室温加热至900℃,并保持1小时以获得金属负载的催化剂。
[0229]
通过icp-ms测量获得的金属负载的催化剂的铂负载量为30重量%。另外,通过icp-ms测量获得的金属负载的催化剂中所含的铂与钴的摩尔比(pt/co比)为7.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0230]
[实施例5]
[0231]
通过与上述实施例4相同的方法获得金属负载的催化剂,不同之处在于使用碳载体e代替碳载体a。通过icp-ms测量获得的所得金属负载的催化剂的铂负载量为30重量%。另外,金属负载的催化剂中所含的铂与钴的摩尔比(pt/co比)为7.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0232]
[实施例6]
[0233]
通过与上述实施例4相同的方法获得金属负载的催化剂,不同之处在于使用碳载体f代替碳载体a。通过icp-ms测量获得的所得金属负载的催化剂的铂负载量为30重量%。另外,金属负载的催化剂中所含的铂与钴的摩尔比(pt/co比)为7.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0234]
[实施例7]
[0235]
通过与上述实施例4相同的方法获得金属负载的催化剂,不同之处在于使用碳载体g代替碳载体a。通过icp-ms测量获得的所得金属负载的催化剂的铂负载量为30重量%。另外,金属负载的催化剂中所含的铂与钴的摩尔比(pt/co比)为7.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0236]
[实施例8]
[0237]
通过与上述实施例4相同的方法获得金属负载的催化剂,不同之处在于使用氯化镍(ii)六水合物(nicl2·
6h2o)代替氯化钴。通过icp-ms测量获得的所得金属负载的催化剂的铂负载量为30重量%。另外,金属负载的催化剂中所含的铂与镍的摩尔比(pt/ni比)为7.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0238]
[实施例9]
[0239]
通过与上述实施例4相同的方法获得金属负载的催化剂,不同之处在于不使用氯化钴。通过icp-ms测量获得的所得金属负载的催化剂的铂负载量为30重量%。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比率为99%或更大。
[0240]
[实施例c1]
[0241]
通过与上述实施例1相同的方法获得金属负载的催化剂,不同之处在于代替在氢气气氛中用毫米波加热,在真空气氛中通过使用高频炉以50℃/min的升温速率进行加热。通过icp-ms测量获得的金属负载的催化剂的铂负载量为20重量%。另外,通过icp-ms测量获得的金属负载的催化剂中所含的铂与铁的摩尔比(pt/fe比)为5.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为1%或更小。
[0242]
[实施例c2]
[0243]
通过与上述实施例1相同的方法获得金属负载的催化剂,不同之处在于使用碳载体d代替碳载体a。通过icp-ms测量获得的金属负载的催化剂的铂负载量为20重量%。另外,通过icp-ms测量获得的金属负载的催化剂中所含的铂与铁的摩尔比(pt/fe比)为5.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0244]
[实施例c3]
[0245]
通过与上述实施例1相同的方法获得金属负载的催化剂,不同之处在于使用碳载体kb代替碳载体a。通过icp-ms测量获得的金属负载的催化剂的铂负载量为20重量%。另外,通过icp-ms测量获得的金属负载的催化剂中所含的铂与铁的摩尔比(pt/fe比)为5.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0246]
[实施例c4]
[0247]
通过与上述实施例1相同的方法获得金属负载的催化剂,不同之处在于使用碳载体v代替碳载体a。通过icp-ms测量获得的金属负载的催化剂的铂负载量为20重量%。另外,
通过icp-ms测量获得的金属负载的催化剂中所含的铂与铁的摩尔比(pt/fe比)为5.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0248]
[实施例c5]
[0249]
通过与上述实施例1相同的方法获得金属负载的催化剂,不同之处在于使用碳载体db代替碳载体a。通过icp-ms测量获得的金属负载的催化剂的铂负载量为20重量%。另外,通过icp-ms测量获得的金属负载的催化剂中所含的铂与铁的摩尔比(pt/fe比)为5.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0250]
[实施例c6]
[0251]
通过与上述实施例1相同的方法获得金属负载的催化剂,不同之处在于使用碳载体m代替碳载体a。通过icp-ms测量获得的金属负载的催化剂的铂负载量为20重量%。另外,通过icp-ms测量获得的金属负载的催化剂中所含的铂与铁的摩尔比(pt/fe比)为5.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0252]
[实施例c7]
[0253]
通过与上述实施例4相同的方法获得金属负载的催化剂,不同之处在于使用碳载体kb代替碳载体a。通过icp-ms测量获得的所得金属负载的催化剂的铂负载量为30重量%。另外,金属负载的催化剂中所含的铂与钴的摩尔比(pt/co比)为7.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0254]
[实施例c8]
[0255]
通过与上述实施例4相同的方法获得金属负载的催化剂,不同之处在于使用碳载体m代替碳载体a。通过icp-ms测量获得的所得金属负载的催化剂的铂负载量为30重量%。另外,金属负载的催化剂中所含的铂与钴的摩尔比(pt/co比)为7.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0256]
[实施例c9]
[0257]
通过与上述实施例1相同的方法获得金属负载的催化剂,不同之处在于使用碳载体kb代替碳载体a并且使用氯化镍(ii)六水合物(nicl2·
6h2o)代替氯化铁。通过icp-ms测量获得的所得金属负载的催化剂的铂负载量为20重量%。另外,金属负载的催化剂中所含的铂与镍的摩尔比(pt/ni比)为7.0。另外,通过xrd获得的金属负载的催化剂中的纯pt的负载量相对于纯pt的负载量和pt合金的负载量之和的比例为10%或更大。
[0258]
[电感耦合等离子体质谱法(icp-ms)]
[0259]
通过icp-ms测量金属负载的催化剂的金属含量。即,首先,将25mg金属负载的催化剂在大气气氛中在800℃下加热并保持3小时以除去金属负载的催化剂中的非金属组分。然后,通过将金属负载的催化剂浸渍在5ml的王水中而使金属负载的催化剂中所含的金属溶解。进一步地,加入蒸馏水以稀释所得物使得总重量变为25g以获得金属溶液。之后,使用顺序型等离子体发射光谱仪(icp-8100,由shimadzu corporation制造)测定所得金属溶液的
pt浓度和过渡金属浓度。
[0260]
然后,将通过将金属溶液的pt浓度(mg/g)乘以金属溶液的重量(25g)而获得的值除以金属负载的催化剂的重量(25mg),并将由此获得的值乘以100以计算金属负载的催化剂的pt含量(重量%)。
[0261]
然后,将通过将金属溶液的过渡金属浓度(mg/g)乘以金属溶液的重量(25g)而获得的值除以金属负载的催化剂的重量(25mg),并将由此获得的值乘以100以计算金属负载的催化剂的过渡金属含量(重量%)。
[0262]
进一步,将通过将pt含量(重量%)除以pt原子量而获得的值除以通过将过渡金属含量(重量%)除以过渡金属原子量而获得的值,从而计算出pt/过渡金属的摩尔比。另外,获得金属负载的催化剂的pt含量(重量%)和过渡金属含量(重量%)的总和作为金属负载的催化剂的金属含量(重量%)。
[0263]
[通过燃烧法的元素分析(chn)]
[0264]
通过金属负载的催化剂的燃烧法进行元素分析。即,通过燃烧法通过使用有机微量元素分析仪(2400ii,perkinelmer co.,ltd.)测量金属负载的催化剂的氮原子含量、碳原子含量和氢原子含量。具体地,在980℃的燃烧管温度和640℃的还原管温度的条件下,通过使用氦气作为载气,对2mg金属负载的催化剂进行分析。
[0265]
然后,将金属负载的催化剂中所含的氮原子、碳原子和氢原子的每个重量除以金属负载的催化剂的重量,并将所得值乘以100,从而计算出金属负载的催化剂的氮原子含量(重量%)、碳原子含量(重量%)和氢原子含量(重量%)。进一步地,将氮原子含量(重量%)除以碳原子含量(重量%)以计算出通过元素分析的n/c比。
[0266]
[x射线光电子能谱法(xps)]
[0267]
通过使用x射线光电子能谱仪(axis nova,由kratos制造),测量金属负载的催化剂的表面上的铂原子、铁原子、钴原子、镍原子、氧原子、氯原子、碳原子和氮原子的每个核能级的光电子能谱。使用alkα射线(10ma,15kv,通能:40ev)作为x射线源。在获得的光电子能谱中,对结合能进行校正,使得源自碳原子的1s轨道的c1s峰的峰顶位于284.5ev。
[0268]
在xps宽扫描分析中,由光电子能谱中的峰面积和检测灵敏度系数确定金属负载的催化剂的表面上的铂原子、碳原子和氮原子的原子浓度(原子%)。另外,将氮原子浓度(原子%)除以碳原子浓度(原子%)以计算出通过xps的n/c比。进一步地,由碳原子和氮原子的原子量比计算氮原子含量(重量%)与碳原子含量(重量%)的比率(xps-n/c重量比)。
[0269]
进一步地,由铂峰面积、检测灵敏度系数和原子量确定通过xps的金属负载的催化剂的铂含量(重量%)。在此,在以下假设下进行计算:金属负载的催化剂不含除了铂原子、铁原子、钴原子、镍原子、氧原子、氯原子、碳原子和氮原子以外的原子。
[0270]
[比表面积、孔体积]
[0271]
通过使用比表面积/孔分布测量装置(tristar 3000,由shimadzu corporation制造)测量金属负载的催化剂的比表面积和孔体积。即,首先,通过将0.1g金属负载的催化剂保持在100℃和6.7
×
10-2
pa下3小时来去除吸附在金属负载的催化剂上的水。然后,通过bet方法由77k下的氮吸附等温线获得金属负载的催化剂的比表面积(m2/g)。通过测量吸附在金属负载的催化剂上的氮量随77k温度下氮气压力变化的变化而获得77k下的氮吸附等温线。
[0272]
同时,根据77k温度下的氮吸附等温线,通过bjh方法获得每个具有0.5nm或更大且2.0nm或更小的直径的孔的体积(cm3/g)、每个具有大于2.0nm且4.0nm或更小的直径的孔的体积(cm3/g),和每个具有大于4.0nm且50.0nm或更小的直径的孔的体积(cm3/g)。bjh方法是barrett、joyner和halenda提出的获得中孔的分布的典型方法(e p barrett,l g joyner and p p halenda,j am chem soc,73,373,(1951))。
[0273]
另外,基于金属负载的催化剂的比表面积和孔体积以及通过icp-ms获得的上述金属含量,还计算出金属负载的催化剂中所含的碳载体的每单位重量的比表面积和孔体积。即,通过将金属负载的催化剂的比表面积和孔体积除以由下式计算的金属负载的催化剂中所含的碳载体的重量比,计算出碳载体的每单位重量的比表面积和孔体积:碳载体的重量比=1-(通过icp-ms获得的金属含量(重量%)/100。
[0274]
[粉末x射线衍射(xrd)]
[0275]
当催化剂含有铂颗粒(纯铂颗粒和/或铂合金颗粒)时,在通过使用cukα射线的粉末x射线衍射获得的粉末x射线衍射图中,铂(111)衍射线出现在衍射角(2θ)在40
°
附近的位置(例如,在36
°
至44
°
的范围内)。
[0276]
在这方面,在包含碳载体和负载在碳载体上的铂颗粒的金属负载的催化剂中,在衍射角(2θ)在40
°
附近的位置处具有峰顶的衍射线出现在x射线衍射图中。衍射线包括至少三种衍射线,即源自纯铂的衍射线、源自铂合金的衍射线和源自碳载体的碳结构的衍射线。
[0277]
这里,源自纯铂的衍射线定义为在衍射角(2θ)为39.6
°
或更大且小于39.8
°
的位置处具有峰顶的衍射线。源自铂合金的衍射线定义为在衍射角(2θ)为39.9
°
或更大且小于43.0
°
的位置处具有峰顶的衍射线。源自碳载体的碳结构的衍射线定义为在衍射角(2θ)为43.3
°
或更大且小于43.7
°
的位置处具有峰顶的衍射线。
[0278]
因此,对于包含碳载体和负载在碳载体上的纯铂颗粒和铂合金颗粒的金属负载的催化剂,在衍射角(2θ)在40
°
附近的位置处具有峰顶的衍射线可以分离成至少三种衍射线。
[0279]
此外,当金属负载的催化剂含有多种具有不同组成和/或晶体结构的铂合金时,会出现多条源自铂合金的衍射线。源自铂合金的衍射线的峰顶所在的衍射角由其组成和晶体结构决定。例如,源自由fept的组成表示的铁铂合金的衍射线定义为在衍射角为41.1
°
或更大且小于41.5
°
的位置处具有峰顶的衍射线。另外,源自由fept3的组成表示的铁铂合金的衍射线定义为在衍射角为40.1
°
或更大且小于40.5
°
的位置处具有峰顶的衍射线。类似地,衍射线的峰顶所在的衍射角例如针对铁铂合金fept7为39.9
°
或更大且小于40.1
°
,针对钴铂合金copt为41.1
°
或更大且小于41.5
°
,针对钴铂合金copt3为40.1
°
或更大且小于40.5
°
,针对钴铂合金copt7为39.9
°
或更大且小于40.1
°
,针对镍铂合金nipt为41.1
°
或更大且小于41.5
°
,针对镍铂合金nipt3为40.1
°
或更大且小于40.5
°
,针对镍铂合金nipt7为39.9
°
或更大且小于40.1
°
。
[0280]
进一步地,当金属负载的催化剂含有多种具有相同组成和晶体结构但微晶直径不同的铂颗粒时,会出现多条衍射线,每条衍射线具有位于相同衍射角位置的峰顶和不同的半峰全宽。
[0281]
因此,当金属负载的催化剂含有多种具有不同组成和/或晶体结构的铂合金时,和/或当金属负载的催化剂含有多种具有相同组成和/或晶体结构但微晶直径不同的铂颗粒时,在通过使用cukα射线获得的x射线衍射图中,在衍射角(2θ)在40
°
附近的位置处具有
峰顶的衍射线包括四种或更多种衍射线。在这种情况下,在衍射角(2θ)在40
°
附近的位置处具有峰顶的衍射线可以分离成四种或更多种衍射线(源自纯铂的衍射线、源自两种或更多种铂合金的衍射线,和源自碳载体的碳结构的衍射线)。
[0282]
现在,将具体描述通过粉末xrd分析金属负载的催化剂的方法。首先,将粉状金属负载的催化剂的样品置于玻璃样品板的凹部(2cm
×
2cm
×
0.5mm(厚度))中并用载玻片按压。因此,样品被均匀地填充到凹部中,使得样品表面和参考表面彼此匹配。然后,将玻璃样品板固定在广角x射线衍射样品台上,使得所填充样品的形状不会塌陷。
[0283]
然后,通过使用x射线衍射仪(rigaku rint2100/pc,rigaku corporation)进行粉末x射线衍射(xrd)测量。施加到x射线管的电压和电流分别设置为50kv和300ma。采样间隔设置为0.1
°
,扫描速度设置为1
°
/min,并且测量角范围(2θ)设置为5
°
至90
°
。cukα用作入射x射线。样品厚度设置为0.5mm,并且发散狭缝宽度β设置为2/3
°
。
[0284]
在获得的xrd图中,铂(111)衍射线出现在衍射角(2θ)在40
°
附近的位置处。然后,首先进行基线校正。即,将连接衍射角(2θ)在35
°
到37
°
附近的衍射强度和衍射角(2θ)在50
°
到52
°
附近的衍射强度的直线确定为基线,并且从衍射线的每个强度中减去基线,从而进行基线校正。
[0285]
接下来,将基线校正后的衍射线分离成源自纯pt的峰、源自一种或多种pt合金的峰和源自碳的峰。通过假设通过分离获得的多个峰中的每一个都用高斯函数来表示并且优化多个峰的高斯函数中的每一个的强度、峰顶的衍射角和半峰全宽,使得关于所有衍射角,通过将衍射线的强度的总和与xrd图的每个衍射角处的多个峰的每个强度的差值(残差)的平方相加而获得的残差平方和变得最小来进行衍射线的分离。
[0286]
这里,将以实施例4的金属负载的催化剂为例描述在衍射角(2θ)在40
°
附近(在36
°
至44
°
的范围内)的位置处具有峰顶的铂(111)衍射线的峰分离。
[0287]
在实施例4的金属负载的催化剂的通过粉末xrd测量获得的xrd图中,在基线校正之后出现在衍射角(2θ)为40.0
°
的位置处具有峰顶的衍射线。该衍射线的上部的形状在宽度方面明显小于下部的形状。因为如此,认为当衍射角(2θ)在40.0
°
附近时,具有较小半峰全宽的第一铂合金的衍射线和具有与第一铂合金不同的微晶直径并且具有大于第一铂合金的半峰全宽的第二铂合金的衍射线重叠。另外,金属负载的催化剂含有碳载体,并且因此在衍射角(2θ)在43.5
°
附近的位置处出现源自碳的衍射线。
[0288]
然后,将具有在40
°
附近的衍射角(2θ)的衍射线分离成由源自第一铂合金的峰、源自第二铂合金的峰、和源自碳的峰形成的三个分量。
[0289]
图1a中示出了峰分离成这三个分量的结果。在图1a中,“基线校正后”的衍射线表示通过对通过xrd测量获得的衍射线进行基线校正而获得的衍射线,并且“合金1”的峰、“合金2”的峰和“碳”的峰分别表示通过“基线校正后”的衍射线的峰分离获得的源自第一铂合金的峰、源自第二铂合金的峰和源自碳的峰。另外,“合金1+2+碳”的峰表示通过将“合金1”的峰、“合金2”的峰和“碳”的峰相加而获得的峰。
[0290]
然而,如图1a所示,当进行基线校正后的衍射线的峰分离使得在37
°
附近的衍射角(2θ)处的尾部扩展和在40
°
附近的峰顶的形状匹配时,39.6
°
和41.0
°
附近的肩部无法再现。
[0291]
在这方面,如上所述,源自纯铂的衍射线在衍射角(2θ)为39.6
°
或更大且小于39.8
°
的位置处具有峰顶,并且源自铂合金的衍射线在衍射角(2θ)为39.9
°
或更大且小于
43.0
°
的位置具有峰顶。因此,认为在基线校正后的衍射线中,源自纯pt的在39.6
°
附近的位置处具有峰顶的衍射线,和源自第三铂合金的在41.0
°
附近的位置处具有峰顶的衍射线是混合的。
[0292]
然后,将具有在40
°
附近的衍射角(2θ)的衍射线分离成由源自纯铂的峰、第一铂合金的峰、源自第二铂合金的峰、源自第三铂合金的峰和源自碳的峰形成的五个分量。
[0293]
图1b中示出了峰分离成这五个分量的结果。在图1b中,“基线校正后”的衍射线表示通过对通过xrd测量获得的衍射线进行基线校正而获得的衍射线,并且“纯pt”的峰、“合金1”的峰、“合金2”的峰、“合金3”的峰和“碳”的峰分别表示通过“基线校正后”的衍射线的峰分离获得的源自纯铂的峰、源自第一铂合金的峰、源自第二铂合金的峰、源自第三铂合金的峰和源自碳的峰。
[0294]
通过将“纯pt”的峰、“合金1”的峰、“合金2”的峰、“合金3”的峰和“碳”的峰相加获得的峰与“基线校正后”的衍射线基本完全匹配,并且因此未在图1b中示出。
[0295]
如上所述,与峰分离为三个分量的情况下的残差平方和相比,峰分离为五个分量的情况下的残差平方和减少,并且变成显著小的值。因此,可以推断出纯铂颗粒、第一铂合金颗粒、第二铂合金颗粒和第三铂合金颗粒作为pt颗粒负载在实施例4的金属负载的催化剂上。
[0296]
然后,通过以下谢乐方程计算纯铂颗粒、第一铂合金颗粒、第二铂合金颗粒和第三铂合金颗粒中的每一个的微晶直径:微晶直径=kλ/βcosθ。这里,在谢乐方程中,k表示谢乐常数(0.94),λ表示cukα射线的波长(0.15418nm),β表示半峰全宽(弧度),θ表示衍射角(弧度)。即,例如,通过将图1b所示的xrd图中的“纯pt”的分离峰的衍射角和半峰全宽代入上述谢乐方程来计算纯铂颗粒的微晶直径。
[0297]
另外,将通过上述峰分离获得的pt分离峰的面积(即“纯pt”的峰面积,“合金1”的峰面积,“合金2”的峰面积和“合金3”的峰面积)各自除以pt分离峰的面积之和来计算pt分离峰中的每一个的峰面积比例。然后,使用这些峰面积比例作为权重,将催化剂金属颗粒的平均微晶直径计算为加权平均值。
[0298]
具体地,在图2中,显示了针对实施例4的金属负载的催化剂计算的“纯pt”、“合金1”、“合金2”和“合金3”的微晶直径和峰面积比例。由以下方程将负载在实施例4的金属负载的催化剂上的pt颗粒(纯pt颗粒和pt合金颗粒)的平均微晶直径计算为5.09nm:平均微晶直径(nm)=(3.9
×
52.3)/100+(24.5
×
3.4)/100+(4.9
×
30.4)/100+(5.2
×
13.9)/100。
[0299]
另外,在“纯pt”、“合金1”、“合金2”和“合金3”中,每个具有5.0nm的微晶直径的pt颗粒的峰面积比例之和计算为每个具有5.0nm或更小的微晶直径的pt颗粒的量相对于pt颗粒的总量的比例(%)。具体地,关于实施例4的金属负载的催化剂,如图2所示,“纯pt”和“合金2”的微晶直径为5.0nm或更小,并且因此“纯pt”和“合金2”的峰面积比例之和82.7%(=52.3%+30.4%)计算为每个具有5.0nm或更小的微晶直径的催化剂金属颗粒的比例。
[0300]
另外,通过铂(111)衍射线的峰分离获得的源自纯pt的分离峰的面积相对于源自纯pt的分离峰的面积和源自一种或更多种pt合金的分离峰的面积之和的比例(%)计算为纯pt的负载量相对于金属负载的催化剂中的纯pt的负载量和pt合金的负载量之和的比例(%)。
[0301]
[每个具有5.0nm或更小的粒径的催化剂金属颗粒的比例(tem观察)]
[0302]
通过使用tem通过以下方法计算每个具有5.0nm或更小的粒径的催化剂金属颗粒的比例(%)。通过使用jeol ltd制造的jem-2010型透射电子显微镜以400,000倍或更大的放大倍数进行金属负载的催化剂的tem观察。即,在所获得的tem图像中,测量100个随机选择的催化剂金属颗粒的最长部分的长度作为粒径。然后,通过将每个具有5.0nm或更小的粒径的催化剂金属颗粒的数量除以100个催化剂金属颗粒的总数所获得的值乘以100,从而计算出每个具有5.0nm或更小的粒径的催化剂金属颗粒的比例(%)。
[0303]
在图3中,示出了实施例4的金属负载的催化剂的tem图像作为用于评估每个具有5.0nm或更小的粒径的催化剂金属颗粒的比例的tem图像的示例。如图3中所示的颗粒之一所示,由箭头指示的最长部分的长度被测量为粒径。
[0304]
[金属负载的催化剂的平均粒径(μm)]
[0305]
测量了包含碳载体和催化剂金属颗粒的金属负载的催化剂的平均粒径。即,通过激光衍射法通过使用纳米粒径分布测量装置(sald-7100h,由shimadzu corporation制造)测量金属负载的催化剂的粒径。
[0306]
具体地,首先将一滴表面活性剂加入10mg金属负载的催化剂中,然后加入40g蒸馏水以制备悬浮液。之后,进行均化器处理20分钟以制备分散体。将制备的分散液滴加到循环有蒸馏水的流动池中,直至衍射/散射光强度的最大值达到50
±
5,并且测定粒径。获得由所得粒径分布(数量分布)确定的中值直径(d50)作为平均粒径。在所得粒径分布(数量分布)的频率(%)为0.001或更大的粒径中,最大值和最小值分别定义为最大粒径和最小粒径。
[0307]
[最外表面上的催化剂金属颗粒的比例]
[0308]
首先,通过使用场发射透射电子显微镜(jem-2100f,由jeol ltd.制造)获取了负载在实施例4、实施例8和实施例c1的金属负载的催化剂中的每一个上的pt颗粒的haadf-stem图像和stem二次电子图像。
[0309]
haadf-stem图像是透射电子图像,因此可以观察到碳载体颗粒内部的金属颗粒,以及碳载体颗粒中的每一个的表面上的金属颗粒。同时,在stem二次电子图像中,只能观察到碳载体的最外表面上的金属颗粒。
[0310]
接下来,进行haadf-stem图像和stem二次电子图像的图像分析。即,通过使用图像处理分析装置(luzex ap,由nireco corporation制造)进行haadf-stem图像的图像分析,并且计算haadf-stem图像的特定视场中的金属颗粒的数量。另外,类似地,进行stem二次电子图像的图像分析,并且计算stem二次电子图像中的与haadf-stem图像的特定视场对应的特定视场内的金属颗粒的数量。然后,通过将stem二次电子图像中的金属颗粒的数量除以haadf-stem图像中的金属颗粒的数量所得的值乘以100,从而计算出金属负载的催化剂的最外表面上的催化剂金属颗粒的比例。
[0311]
在图4a和图4b中,分别示出了针对实施例4的金属负载的催化剂获取的haadf-stem图像和stem二次电子图像。如图4a所示,在haadf-stem图像中,观察到存在于碳载体的表面上和碳载体的内部的金属颗粒。同时,如图4b所示,在stem二次电子图像中,仅观察到存在于碳载体的表面上的金属颗粒。
[0312]
图4a中虚线所包围的视场中的金属颗粒的数量为1,219。另外,图4b中虚线所包围的视场内的金属颗粒的数量为125。因此,在实施例4的金属负载的催化剂中,在最外表面上的催化剂金属颗粒的比例计算为约10%。类似地,在金属负载的催化剂的最外表面上的催
化剂金属颗粒的比例在实施例8中为约4%,并且在实施例c1中为约13%。
[0313]
[拉曼光谱法]
[0314]
通过拉曼光谱法分析金属负载的催化剂。通过使用horiba微型激光拉曼光谱测量装置(labram,horiba jobin yvon)测量拉曼光谱。用于测量的激光具有532nm的激发波长和50mw的输出,并且通过中性密度滤光片d3在曝光90秒
×
积分2次的条件下进行测量,以得到拉曼光谱。
[0315]
对所获得的拉曼光谱进行基线校正。即,将连接拉曼位移(cm-1
)在800cm-1
附近的散射强度和拉曼位移(cm-1
)在2,000cm-1
附近的散射强度的直线确定为基线,并且从散射光谱的每个强度中减去基线,从而进行基线校正。
[0316]
接下来,识别了在1,600cm-1
附近具有峰顶的g带和在1,360cm-1
附近具有峰顶的d带。此外,基于g带的强度ig(g带的峰顶强度)、d带的强度id(d带的峰顶强度)以及g带和d带之间的最小强度iv,获得了g带的半峰全宽(cm-1
)、d带的半峰全宽(cm-1
)和iv/ig比。
[0317]
在图5中,示出了通过分析在实施例1中获得的金属负载的催化剂的通过拉曼光谱法获得的拉曼光谱而获得的结果作为上述拉曼光谱的一个示例。在图5中,横轴表示拉曼位移(cm-1
),纵轴表示散射强度,虚线表示基线,ad表示与d带的峰顶相对应的拉曼位移(cm-1
),bd表示与在ad的低波数侧上表现出d带的强度id的一半的强度的拉曼光谱相对应的拉曼位移(cm-1
),ag表示与g带的峰顶相对应的拉曼位移(cm-1
),并且bg表示与在ag的高波数侧上表现出g带的强度ig的一半的强度的拉曼光谱相对应的拉曼位移(cm-1
)。
[0318]
[程序升温脱附法]
[0319]
将金属负载的催化剂安装在程序升温脱附装置(由microtracbel corp.制造)中,并以20ml/min的速度循环载气(he)以加热碳催化剂。用四极质谱仪(qms)测量脱附气体。
[0320]
具体地,首先,对金属负载的催化剂进行预处理(通过热处理使催化剂表面官能团脱附)。即,首先,将0.05g的金属负载的催化剂填充到由石英制成的反应管的中央部,并设置在程序升温脱附装置中。通过在氮气气氛中以10℃/min的升温速率将反应管加热至600℃并将反应管保持在600℃下30分钟来进行预处理。
[0321]
接下来,使测量气氛稳定化。即,通过在氦气(he)气氛中将反应管保持在25℃下120分钟而使装置内的气氛稳定化。之后,对金属负载的催化剂进行加热处理,并且测量600℃至1000℃的温度范围和800℃至1000℃的温度范围中的每一个中的氮脱附量。即,在上述稳定后,将金属负载的催化剂再次加热并以10℃/min的升温速率升温至1,000℃,从而进行金属负载的催化剂的热处理,并且测量其表面上的氮官能团的量。
[0322]
更具体地,对金属负载的催化剂进行加热处理,并测量脱附的氮气(n2)的量。即,在装置内的气氛稳定后,以10℃/分钟的升温速率将反应管升温至1000℃。在该升温至1,000℃期间,当以20ml/min循环氦(he)气时,使用质量数14检测出通过使含氮化合物脱附产生的氮(当使用质量数28时,除了n2之外,还含有气体诸如co和c2h4(特别地,主要含有co),并且因此使用质量数14)。首先,对得到的光谱进行基线校正,此后记录温度(横轴)与检测强度(纵轴)的相关性。
[0323]
然后,计算600℃至1000℃的温度范围和800℃至1000℃的温度范围中的每一个中的氮的检测强度(检测强度面积)的积分值,从而确定在温度范围中的每一个内脱附的氮的释放量。
[0324]
同时,通过使用氮气作为标准气体,创建了显示氮气的释放量与检测强度面积之间的相关性的校准曲线。然后,将基于通过测量获得的检测强度面积和校准曲线量化的值除以用于测量的碳催化剂的量,从而确定从金属负载的催化剂的氮脱附量(金属负载的催化剂的每单位重量的脱附氮气的释放量)(mоl/g)。
[0325]
另外,基于上述获得的金属负载的催化剂的每单位重量的氮脱附量(mol/g)和通过icp-ms得到的上述金属含量(重量%),还计算出金属负载的催化剂中所含的碳载体的每单位重量的氮脱附量(mоl/g-载体)。即,将从金属负载的催化剂的氮脱附量除以通过下式计算的金属负载的催化剂中所含的碳载体的重量比,从而计算出碳载体的每单位重量的氮脱附量(mоl/g-载体):碳载体重量比=1-(通过icp-ms获得的金属含量(重量%))/100。
[0326]
[有效铂催化剂表面积(ecsa)]
[0327]
通过使用旋转圆盘电极测量方法对如上所述制备的金属负载的催化剂进行循环伏安法(cv)测量。首先,将通过将5.0mg金属负载的催化剂、蒸馏水和8∶2重量比的异丙醇混合而制备的500μl水溶液和50μl电解质树脂溶液(de2020cs,由dupont制造,电解质树脂浓度:20重量%)混合。将获得的悬浮液进行超声波分散处理5分钟,然后使用均化器进一步进行分散处理2分钟,从而获得催化剂墨水。
[0328]
接下来,将所获得催化剂墨水施加于旋转圆盘电极,使得金属负载的催化剂的负载量(催化剂金属的负载量和碳载体的负载量之和)变成0.1mg/cm2,并且用干燥器干燥以形成催化剂层。然后,将其上形成有催化剂层的旋转圆盘电极安装在电化学电池(由bas inc.制造)中。在电化学电池中,使用可逆氢电极(rhe)作为参比电极,并且使用0.1m的高氯酸水溶液作为电解液。将电解液用氮气饱和10分钟,并且以0.05v/sec的扫描速度从0.06v的低电位到1.2v的高电位进行5个循环的电位扫描。
[0329]
在所获得循环中的稳定的第5个循环中阴极扫描时的电流值中,从在循环伏安图的0.06v至0.40v的电位范围内的还原电流值的电量中减去基于0.40v时的还原电流值的由在0.06v至0.40v的电位范围内的非法拉第电流引起的电量,从而获得氢吸附电量(单位:c)。
[0330]
将氢吸附电量除以相对于铂的氢吸附的理论面积等效电量(2.10c/m2),并进一步除以铂的重量(g),从而计算氢吸附中的有效铂催化剂表面积(m2/g-铂)(以下称为“h
2-ecsa(电化学表面积)”)。
[0331]
同时,通过使用旋转圆盘电极进行co溶出伏安法测量。首先,通过与上述cv测量相同的方法制作旋转圆盘电极,并安装在电化学电池中。也以与上述cv相同的方式制备参比电极和电解液。
[0332]
然后,进行预处理。即,将电解液用氮气饱和10分钟,并且以0.1v/sec的扫描速度从0.06v的低电位到1.2v的高电位进行20个循环的电位扫描。
[0333]
在完成上述预处理后,通过与上述cv测量相同的方法进行cv测量。接下来,将0.3%的co气体饱和5分钟,并且进一步地,在将旋转圆盘电极以1,600rpm旋转时,以0.1v/sec的扫描速度从0.06v的低电位到1.2v的高电位进行5个循环的电位扫描。
[0334]
此后,施加0.05v的电压,并且在以1,600rpm旋转的同时将旋转圆盘电极保持40分钟。在将电解液用氮气饱和另外10分钟时施加0.05v的电压,并且将旋转圆盘电极以1,600rpm旋转。然后,以0.05v/sec的扫描速度从0.06v的低电位到1.2v的高电位进行5个循环
的电位扫描。
[0335]
在如此获得第一循环中的阳极扫描时的电流值中,从在循环伏安图的0.6v至1.1v的电位范围内的还原电流值的电量中减去基于0.6v时的还原电流值的由在0.6v至1.1v的电位范围内的非法拉第电流引起的电量,从而获得co吸附电量(单位:c)。
[0336]
将co吸附电量除以相对于铂的co吸附的理论面积等效电量(4.20c/m2),并进一步除以铂的重量(g),从而计算co吸附中的有效铂催化剂表面积(m2/g-铂)(以下称为“co-ecsa”)。
[0337]
[非铂活性位点的pt当量]
[0338]
首先,通过使用市售的铂催化剂(unpc40-ii,由ishifuku metal industry co.,ltd.制造),制造了其中每单位阴极面积的铂负载量为0.012mg/cm2、0.028mg/cm2、0.045mg/cm2、0.067mg/cm2和0.100mg/cm2的五个膜电极组件(mea)。铂催化剂由作为载体的炭黑和负载在载体上的39重量%铂颗粒形成。
[0339]
接下来,通过使用多个mea中的每一个进行发电测试。即,分别向阳极侧和阴极侧供应氢气和氧气,并在75℃的电池温度和100%rh的相对湿度的条件下扫描电流,从而获得0.1a/cm2电流密度下的电压(mv)和mea的铂负载量之间的相关性。结果,使用具有0.012mg/cm2的铂负载量的mea时的电压比使用具有0.1mg/cm2的铂负载量的mea时的电压(以下称为“基准电压”)低120mv。
[0340]
同时,代替上述市售铂催化剂,制备了含有不负载催化剂金属颗粒的碳载体的mea。以与上述使用市售铂催化剂的情况相同的方式进行发电测试,并且测量0.1a/cm2的电流密度下的电压(mv)。
[0341]
具体地,例如,在使用含有0.3mg/cm2的量的碳载体a的mea的发电测试中,0.1a/cm2的电流密度下的电压(mv)比上述参考电压低120mv。因此,认识到0.3mg/cm2的量的碳载体a具有与0.012mg/cm2的量的铂催化剂相对应的催化活性。即,0.1mg/cm2的量的碳载体a被评价为具有与0.004mg/cm2的量的铂催化剂相对应的催化活性。
[0342]
这里,例如,当在铂含量变为0.1mg/cm2,阴极催化剂层中碳载体a的含量(金属负载的催化剂中的含量:80重量%)变为0.4mg/cm2,并且金属负载的催化剂的含量变为0.5mg/cm2的这种量的上述发电测试的阴极催化剂层中,使用实施例1的金属负载的催化剂(铂颗粒的含量:20重量%,碳载体a的含量:80重量%)代替上述市售铂催化剂时。
[0343]
如上所述,0.1mg/cm2的量的碳载体a具有与0.004mg/cm2的量的铂催化剂相对应的催化活性。因此,包含0.4mg/cm2量的碳载体a的阴极催化剂层被评价为具有与0.016mg/cm2的量的铂催化剂相对应的催化活性。然后,将具有与0.4mg/cm2的量的碳载体a相对应的催化活性的0.016mg/cm2的量的铂催化剂除以碳载体a的负载量(0.0004g/cm2),从而计算出40.0(mg-pt/g-载体)的碳载体a中非铂活性位点的铂当量。
[0344]
[初始性能和耐久性]
[0345]
制造了具有催化剂层的电池阴极,该催化剂层包含在其上形成的金属负载的催化剂。即,首先,将与碳载体的重量比为0.9的这种量的电解质(ew700)加入到0.25g的金属负载的催化剂中,并且添加各2g的蒸馏水和1-丙醇以制备电解质溶液。将电解质溶液和25g球装入罐中并用球磨机以200rpm混合50分钟,从而获得包含均匀分散的金属负载的催化剂的用于催化剂层的浆状组合物。
[0346]
将获得的用于催化剂层的浆状组合物施加到的气体扩散层(“29bc”,由sgl carbon japan co.,ltd.制造)的面积为5cm2的区域上(2.3cm
×
2.3cm)使得每单位面积电池电极的金属负载的催化剂的含量变为0.1mg/cm2,并且干燥以在气体扩散层上形成催化剂层。以此方式,获得了具有催化剂层的电池电极,该催化剂层包含在其上形成的金属负载的催化剂。
[0347]
接下来,制造包括电池电极的燃料电池,该电池电极具有含有在其上形成的金属负载的催化剂的催化剂层。即,作为正极,使用包括如上所述制备的催化剂层(正电极催化剂层)的电池电极。
[0348]
同时,如下所述制造负电极。将0.5g pt/c(unpc40-ii,由ishifuku metal industry co.,ltd.制造)、10g 5%nafion(商标)、2g蒸馏水和25g球装入罐中并用球磨机以200rpm混合50分钟,从而制备浆状pt/c组合物。除了将浆状pt/c组合物施加到气体扩散层(5cm2)上之外,以与上述正电极相同的方式制造包括由pt/c组合物形成的催化剂层(负电极催化剂层)的负电极,使得每单位面积施加的pt/c的量变为0.1mg/cm2。
[0349]
然后,在上述正电极催化剂层和上述负电极催化剂层之间设置固体聚合物电解质膜(“nafion(商标)211”,由dupont制造),并且将所得物在150℃和1mpa的条件下压力结合3分钟,从而制造mea。将一对垫片粘合到mea上,并且进一步将所得mea夹在一对隔板之间,从而制造燃料电池单元电池。
[0350]
之后,将如上所述制造的单元电池安装在燃料电池自动评估系统(由toyo corporation制造)中。首先进行发电测试,然后进行耐久性测试。
[0351]
在发电测试中,在35kpa的背压下,将饱和的加湿空气(氧气)以2.5l/min(相对湿度:100%)供应到单元电池的正电极侧,并且将饱和的加湿氢气以1.0l/min(相对湿度:100%)供应到单元电池的负电极侧。电池温度设置为75℃,并且测量开路电压5分钟。之后,将电池电流密度在从3.0a/cm2到0a/cm2的每个电流密度下保持3分钟,并测量电池电压。在发电测试中,将在0.2a/cm2的电流密度下观察到的电压(mv)记录为表现出金属负载的催化剂的初始催化活性的指标之一。
[0352]
此外,针对每个电位计算由电位和电流密度获得的输出密度,并将最高值测量为最大输出密度(mw/cm2)。另外,记录耐久性测试开始时1.0a/cm2下的电压(mv)。
[0353]
之后,将电池温度设置为75℃。在35kpa的背压下,将饱和的加湿氮气以0.5l/min(相对湿度:100%)供应到单元电池的两侧,并且将饱和的加湿氢气以0.5l/min(相对湿度:100%)供应到单元电池的阳极侧。通过重复将电位在0.6v下保持30秒和在1.0v下保持60秒的矩形波循环来进行耐久性测试。
[0354]
在上述矩形波循环重复2,100次后,再次进行发电测试。记录耐久性测试后1.0a/cm2下的电压(mv)。然后,将从在耐久性测试之前的发电测试中作为初始性能测量的上述电压(mv)中减去在耐久性测试后的发电测试中测量的上述电压(mv)(2,100个循环后的电压(mv))获得的值作为2100次循环后的电压下降量(mv)。
[0355]
[结果]
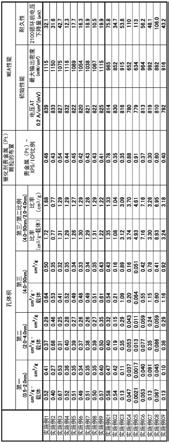
[0356]
在图6a、图6b和图6c中,示出了通过评估每个实施例的金属负载的催化剂的特性而获得的结果。如图6a所示,使用实施例1至9的金属负载的催化剂的mea中的每一个的初始性能和耐久性优于实施例c1至c9的初始性能和耐久性。例如,使用实施例c1、c4、c5和c8的
金属负载的催化剂的mea的耐久性显著较差。此外,使用实施例c2的金属负载的催化剂的mea具有低的最大输出密度。在使用实施例c3至c9的金属负载的催化剂的mea中,0.2a/cm2的电流密度下的电压、最大输出密度和耐久性中的两个或更多个显著低。
[0357]
如图6a所示,实施例1至9的金属负载的催化剂的第一孔的体积大于实施例c3至c9的第一孔的体积。另外,实施例1至9的金属负载的催化剂的第二孔和第三孔的体积大于实施例c2、c4至c6和c8的第二孔和第三孔的体积。
[0358]
然而,实施例c3、c7和c9的第三孔的体积显著大于实施例1至9的金属负载的催化剂的第三孔的体积。因为如此,实施例1至9的金属负载的催化剂的第三孔/第二孔体积比小于实施例c3至c9的第三孔/第二孔体积比。
[0359]
另外,如图6a所示,实施例c1、c4和c5的金属负载的催化剂的贵金属(pt)-xps/icp比显著大于实施例1至9的贵金属(pt)-xps/icp比。即,在实施例c1、c4、c5的金属负载的催化剂中,在碳载体的表面附近负载了相对大量的pt颗粒。
[0360]
如图6b所示,实施例1至9的金属负载的催化剂的chn-n/c比和xps-n/c比显著大于实施例c3至c9的chn-n/c比和xps-n/c比。另外,实施例1至9的金属负载的催化剂的tpd中的氮脱附量显著大于实施例c2至c9的氮脱附量。
[0361]
如图6c所示,实施例1至9的金属负载的催化剂的通过xrd获得的催化剂金属(pt)颗粒的平均微晶直径显著小于实施例c2、c4至c6、c8的平均微晶直径。另外,实施例1至9的金属负载的催化剂的通过tem观察获得的每个具有5.0nm或更小的粒径的催化剂金属(pt)颗粒的比例显著大于实施例c1至c9的比例。当催化剂金属的初级颗粒聚集形成次级颗粒时,在xrd中评估对应于初级颗粒的微晶直径,而在tem观察中评估次级颗粒的粒径。因此,通过xrd获得的微晶直径和通过tem观察获得的粒径不一定相互匹配。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1