一种用于制备羟基芳酮的催化剂及其制备方法和应用与流程
1.本发明属于羟基芳酮的制备领域,具体涉及一种用于制备羟基芳酮的催化剂及其制备方法和应用。
背景技术:
2.羟基芳酮分子中含有苯环上的羟基、酮基,是一种非常重要的精细化学品和合成中间体,在医药、化妆品、食品、合成材料等领域具有广泛的应用。其中,对羟基苯乙酮为例,它是一种天然植物提取物,天然存在于菊科植物滨蒿的茎、叶,茵陈蒿、萝藦科植物、人参娃儿藤等植物的根中。其在医药、染料、化妆品、液晶材料、合成树脂等领域具有重要的应用价值。邻羟基苯乙酮主要用作医药中间体及香料,如ia类抗心律失常药物盐酸普罗帕酮(propafenone hydrochloride)、抗真菌类药盐酸氯康哇(croconazole hydrochloride)等的重要中间体。
3.羟基苯乙酮的合成方法主要有醋酸苯酯法、对氨基苯乙酮法、光催化法等。其中,醋酸苯酯法是醋酸苯酯通过分子内重排得到羟基苯乙酮,原子利用率很高,因此备受亲睐,其有效的催化体系为lewis酸类、酸类和离子液体类催化剂,但是存在对位羟基芳酮的选择性低的缺点。
4.金属有机骨架材料是一类具有本征双亲性结构的聚合物晶体材料。mofs结构中的极性节点可通过配位作用、离子交换、静电作用等多种作用力与分子之间发生强烈地相互作用;并且,mofs具有丰富的孔隙结构特点,有利于通过原位包埋、后期修饰等多种方式制备复合材料以改善其应用性能。
技术实现要素:
5.本发明针对现有技术中在取代的烷基酚酯或卤代酚酯催化合成羟基芳酮中酚酯转化率和羟基芳酮选择性低的问题,提供了一种新的用于制备羟基芳酮的催化剂。该催化剂具有提高酚酯转化率和羟基芳酮选择性的特点。
6.为此,本发明第一方面提供了一种用于制备羟基芳酮的催化剂,其包括有机金属骨架材料和苄基功能化的离子液体;且所述催化剂具有基本上如下的x-射线衍射图谱;
[0007][0008]
其中,(a)=
±
0.3
°
。
[0009]
本发明所述催化剂中的有机金属骨架材料和苄基功能化的离子液体的含量即为合成所述催化剂时采用的有机金属骨架材料和苄基功能化的离子液体的实际用量。由于所述催化剂中的苄基功能化的离子液体是被限域在有机金属骨架材料的孔道中,多余的未在孔道中反应的苄基功能化的离子液体会被溶剂洗掉,因此本发明对催化剂中的有机金属骨架材料和苄基功能化的离子液体的含量没有明确限定,本领域技术人员可以根据实际需要进行调整。
[0010]
在本发明的一些实施方式中,所述有机金属骨架材料选自irmof、zifs、pcps和mil中的一种或多种,优选为mil。
[0011]
本发明中,pcps(多孔配位聚合物)是一类通过配位键将金属团簇(或离子)和有机
配体连接起来的高结晶多孔杂化材料。
[0012]
在本发明的一些优选的实施方式中,所述mil选自mil-53、mil-100和mil-101中的一种或多种,优选为mil-101,更优选为mil-101(cr)。
[0013]
在本发明的一些实施方式中,所述苄基功能化的离子液体的结构中含有由氮杂环有机化合物和苄卤类化合物所形成的阳离子以及金属卤化物。
[0014]
在本发明的一些实施方式中,所述氮杂环有机化合物选自咪唑、烷基咪唑、吡啶、烷基吡啶、嘧啶、烷基嘧啶、吡咯和烷基吡咯中的一种或多种;优选地,所述烷基选自c1~c5的烷基中的至少一种;进一步优选地,所述氮杂环有机化合物选自吡啶、4-甲基吡啶、3-甲基吡啶和3、5-二甲基吡啶中的一种或多种。
[0015]
本发明第二方面提供了一种如本发明第一方面所述催化剂的制备方法,其包括以下步骤:
[0016]
s1,将溶剂a、有机金属骨架材料、氮杂环有机化合物和苄卤类化合物混合后进行反应,然后经冷却、过滤、洗涤和干燥,获得固体产品;
[0017]
s2,将溶剂b、所述固体产品与lewis酸混合后进行反应,然后经冷却、过滤、洗涤和干燥,获得所述催化剂。
[0018]
在本发明的一些实施方式中,所述氮杂环有机化合物、苄卤类化合物与lewis酸的摩尔比为(0.5~1.5):(0.5~1.5):(1.5~2.5);优选为(0.8~1.2):(0.8~1.2):(1.8~2.2);进一步优选为1:1:2。
[0019]
本发明对上述反应中有机金属骨架材料中的用量没有明确限定,本领域技术人员可以根据实际需要进行调整。
[0020]
在本发明的一些实施方式中,所述氮杂环有机化合物选自咪唑、烷基咪唑、吡啶、烷基吡啶、嘧啶、烷基嘧啶、吡咯和烷基吡咯中的一种或多种;优选地,所述烷基选自c1~c5的烷基中的至少一种;进一步优选地,所述氮杂环有机化合物选自吡啶、4-甲基吡啶、3-甲基吡啶和3、5-二甲基吡啶的一种或多种。
[0021]
在本发明的另一些实施方式中,所述苄卤类化合物选自氯苄、2,4,6-三甲基苄氯、对三氟甲基苄氯、苄溴和4-异丙基苄溴中的一种或多种。
[0022]
在本发明的一些实施方式中,所述lewis酸选自三氯化铝、三溴化铝、三氯化铁、氯化锌、三氟化硼、氯化镓、氯化铟、氯化亚铜中的一种或多种;优选选自三氯化铝和三氯化铁中的一种或多种。
[0023]
在本发明的另一些实施方式中,所述溶剂a和溶剂b各自独立地选自氯苯、硝基苯、甲苯、硝基甲烷、乙腈、二氯甲烷中的任意一种。
[0024]
本发明对溶剂a和溶剂b的用量没有明确限定。一般地,只要能溶解相应的溶质即可。在本发明的一些具体实施方式中,上述溶剂的用量使得相应溶质(溶剂所需溶解的溶质)的浓度为0.001mol/l~饱和溶解度。
[0025]
本发明中,所述有机金属骨架材料在使用前可以进行预干燥。
[0026]
在本发明的另一些实施方式中,步骤s1中,所述反应的温度为0~100℃,优选为30~40℃;和/或,所述反应的时间为2~72h,优选为12~15h。
[0027]
在本发明的一些实施方式中,步骤s2中,所述反应的温度为0~100℃,优选为25~35℃;和/或,所述反应的时间为2~20h,优选为8~15h。
[0028]
本发明所述方法将苄基功能化的离子液体原位限域在mil-101(cr)的孔道中,进而提高了苄基功能化的离子液体的性能。由本发明所述方法制得的催化剂具有提高酚酯转化率和羟基芳酮选择性的特点。
[0029]
本发明第三方面提供了一种由烷基酚酯或卤代酚酯制备羟基芳酮的方法,其包括在溶剂c或非溶剂状态下,采用如本发明第一方面所述的催化剂或第二方方面所述方法所制备的催化剂催化烷基酚酯或卤代酚酯进行分子内重排,进而合成羟基芳酮。
[0030]
在本发明的一些实施方式中,所述烷基苯酚或卤代苯酚具有如化学式a或化学式b所示的结构式;
[0031]
化学式a;
[0032]
化学式b;
[0033]
其中,所述r2和r3各自独立任选自h或c1~c6的烷基;所述r1和r4各自独立任选自c1~c6的烷基或c6~c9的芳基;所述x选自f、cl、br和i中的任意一种。
[0034]
在本发明的另一些实施方式中,所述烷基酚酯或卤代酚酯与所述催化剂的重量比为1:(0.001~20);优选为1:(0.01~5)。
[0035]
在本发明的一些实施方式中,所述溶剂c选自氯苯、硝基苯、甲苯、硝基甲烷、乙腈和二氯甲烷中的任意一种。
[0036]
在本发明的另一些实施方式中,所述催化的条件包括:温度为20~200℃,优选为20~100℃;和/或,压力为常压~6mpa,优选为常压~2mpa;时间为0.1~10h,优选为0.5~5h。
[0037]
本发明的有益效果为:本发明所述催化剂中的苄基功能化的离子液体被原位限域在有机金属骨架材料(如,mil-101(cr))的孔道中,进而提高了苄基功能化的离子液体的性能。将本发明所述方法制得的催化剂应用到羟基芳酮的制备中,具有提高酚酯转化率和羟基芳酮选择性的特点。
附图说明
[0038]
下面将结合附图对本发明作进一步说明。
[0039]
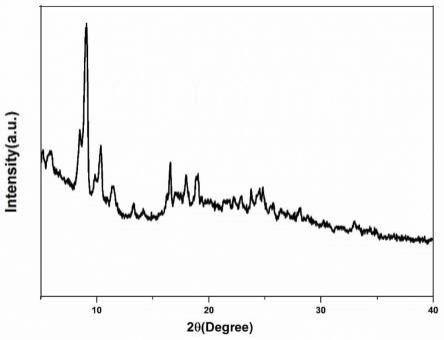
图1为本发明实施例1中所采用的有机金属骨架材料mil-101(cr)的xrd谱图。
[0040]
图2为本发明实施例1中所制备的催化剂mil-101(cr)-fe-1的xrd谱图。
[0041]
图3为本发明实施例1中所采用的有机金属骨架材料mil-101(cr)的ir谱图,主要峰位置为:3441cm-1
、1653cm-1
、1617cm-1
、1505cm-1
、1384cm-1
、1106cm-1
、749cm-1
。
[0042]
图4为本发明实施例1中所制备的有机金属催化剂mil-101(cr)-fe-1的ir谱图,主要峰位置为:3407cm-1
、3053cm-1
、1702cm-1
、1651cm-1
、1622cm-1
、1486cm-1
、1398cm-1
、1300cm-1
、1157cm-1
、1105cm-1
、1016cm-1
、749cm-1
、680cm-1
;其中在3053cm-1
为苄基的峰,1702cm-1
为
苯环的峰,1398cm-1
、1157cm-1
、680cm-1
为吡啶环的峰,1300cm-1
、1016cm-1
为苄基苯环的峰。
具体实施方式
[0043]
为使本发明更加容易理解,下面将结合实施例来进一步详细说明本发明,这些实施例仅起说明性作用,并不局限于本发明的应用范围。本发明中所使用的原料或组分若无特殊说明均可以通过商业途径或常规方法制得。
[0044]
【制备例1】
[0045]
将mil-101(cr)(xrd谱图如图1所示,ir谱图如图3所示)置于150℃烘箱中干燥过夜。
[0046]
在干燥后的100ml三口烧瓶中加入25ml无水乙腈,2g干燥后的mil-101(cr),0.25ml吡啶,搅拌下滴加0.32ml苄氯,室温反应12小时。将体系过滤,无水乙腈充分洗涤滤饼,真空干燥,获得固体产品。
[0047]
将固体产品加入干燥后的100ml三口烧瓶中,加入25ml无水乙腈,搅拌下加入1g无水三氯化铁,室温反应12小时。分出乙腈层,再用无水乙腈充分洗涤,真空干燥,即得催化剂mil-101(cr)-fe-1(xrd谱图如图2所示,ir谱图如图4所示)。
[0048]
【制备例2】
[0049]
将mil-101(cr)置于150℃烘箱中干燥过夜。
[0050]
在干燥后的100ml三口烧瓶中加入25ml无水乙腈,2g干燥后的mil-101(cr),0.25ml吡啶,搅拌下滴加0.32ml苄氯,室温反应12小时。将体系过滤,无水乙腈充分洗涤滤饼,真空干燥,获得固体产品。
[0051]
将固体产品加入干燥后的100ml三口烧瓶中,加入25ml无水乙腈,搅拌下加入0.82g氯化铝,室温反应12小时。分出乙腈层,再用无水乙腈充分洗涤,真空干燥,即得催化剂mil-101(cr)-al-1。
[0052]
【制备例3】
[0053]
将mil-101(cr)置于150℃烘箱中干燥过夜。
[0054]
在干燥后的100ml三口烧瓶中加入25ml无水乙腈,2g干燥后的mil-101(cr),0.25ml吡啶,搅拌下滴加0.32ml苄氯,室温反应12小时。将体系过滤,无水乙腈充分洗涤滤饼,真空干燥,获得固体产品。
[0055]
将固体产品加入干燥后的100ml三口烧瓶中,加入25ml无水乙腈,搅拌下加入0.84g氯化锌,室温反应12小时。分出乙腈层,再用无水乙腈充分洗涤,真空干燥,即得催化剂mil-101(cr)-zn-1。
[0056]
【制备例4】
[0057]
将mil-101(cr)置于150℃烘箱中干燥过夜。
[0058]
在干燥后的100ml三口烧瓶中加入25ml无水乙腈,2g干燥后的mil-101(cr),0.25ml吡啶,搅拌下滴加0.41ml对三氟甲基苄氯,室温反应12小时。将体系过滤,无水乙腈充分洗涤滤饼,真空干燥,获得固体产品。
[0059]
将固体产品加入干燥后的100ml三口烧瓶中,加入25ml无水乙腈,搅拌下加入1g无水三氯化铁,室温反应12小时。分出乙腈层,再用无水乙腈充分洗涤,真空干燥,即得催化剂mil-101(cr)-fe-2。
1,将体系加热至80℃保持4小时,冷却至室温。加入50ml水充分搅拌后用乙酸乙酯萃取三次,合并有机相,无水na2so4干燥后过滤,减压蒸除溶剂,残余物经gc分析。醋酸苯酯的转化率为98.5%,羟基苯乙酮的选择性为90.3%。
[0078]
应当注意的是,以上所述的实施例仅用于解释本发明,并不构成对本发明的任何限制。通过参照典型实施例对本发明进行了描述,但应当理解为其中所用的词语为描述性和解释性词汇,而不是限定性词汇。可以按规定在本发明权利要求的范围内对本发明作出修改,以及在不背离本发明的范围和精神内对本发明进行修订。尽管其中描述的本发明涉及特定的方法、材料和实施例,但是并不意味着本发明限于其中公开的特定例,相反,本发明可扩展至其他所有具有相同功能的方法和应用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1