一种膦胺锰配合物催化剂及其制备方法和应用
1.本发明涉及羰基衍生物加氢催化剂技术领域,尤其涉及一种膦胺锰配合物催化剂及其制备方法和应用。
背景技术:
2.催化醛、酮、酯等羰基衍生物加氢制醇是工业上极为重要的化学转化过程。例如,目前全世界每年通过催化酯类化合物加氢所制备的醇高达数百万吨。制备可高效催化羰基衍生物加氢的催化剂是实现这一转化过程的关键。
3.相比于负载型多相催化剂多需要较为苛刻的反应条件(473-573k,140
‑ꢀ
300barh2),均相催化剂在温和的反应条件下即可表现出优异的加氢活性,有一定的优势。1995年,noyori等首次报道发现ru(ii)-phosphine与乙二胺配体构成的配合物催化剂在均相条件下可有效催化醛类和酮类分子加氢(j.am.chem. soc.,1995,117,2675
–
2676);2006年,saudan等成功将膦胺配体构成的钌配合物催化剂应用于酯类分子的均相加氢反应研究中(国际专利wo 2006106483 和wo 2006106484)。
4.近十年来,基于ru、os、ir等贵金属,研究者进一步发展制备了一系列不同结构的均相加氢催化剂。然而,贵金属的使用所带来的成本问题和环境问题已经引起各方面的广泛关注,亟待开发基于非贵金属的加氢催化剂。
技术实现要素:
5.针对现有技术中缺陷与不足的问题,本发明提出了一种膦胺锰配合物催化剂及其制备方法和应用,可用于醛、酮、酯、酰胺等多种不同结构的羰基衍生物催化加氢制醇,相比于贵金属催化剂,该催化剂的开发利用具有成本低廉和环境友好等多方面优点。
6.本发明解决其技术问题所采用的技术方案是:
7.一种膦胺锰配合物催化剂,所述配合物催化剂配位有双齿膦胺配体或四齿膦胺配体,其结构式如下:
8.9.上述结构中,
10.r为苯基、五氟苯基、噻吩基、异丙基、叔丁基、硅三甲基或呋喃亚甲基;
11.r
′
、r
″
为氢、甲基、乙基、异丙基、叔丁基或苄基;
12.l5、l6为羰基、氮杂环卡宾、氢或乙腈;
13.金属mn的价态为+1或+2;
14.x为负一价的阴离子,其可选自不限于卤素离子、硼氢根离子、氢氧根离子、腈根离子或硫腈根离子中的任意一种;
15.n的数值为0或1或2。
16.进一步的,所述结构式中r优选苯基或异丙基,r
′
、r
″
优选氢或甲基, l5、l6优选羰基或氮杂环卡宾。
17.一种膦胺锰配合物催化剂的制备方法,在绝氧绝水的条件下采用标准 schlenk操作技术,在室温到回流的温度范围内,将膦胺配体l1(或l2、l3、 l4)与金属mn化合物按照1:0.5~4的比例在有机溶剂中反应2~100小时得到配合物催化剂。
[0018][0019]
进一步的,所述有机溶剂可采用不限于甲苯、四氢呋喃、1,4-二氧六环、乙醇、异丙醇、二氯甲烷或乙醚中的一种。
[0020]
进一步的,所述有机溶剂优选甲苯或四氢呋喃。
[0021]
进一步的,一种膦胺锰配合物催化剂的应用,应用于羰基衍生物分子催化加氢反应,具体反应步骤如下:在氢气气氛下,起始氢气压力在10~100bar范围内,在20~160℃的温度条件下,催化剂与底物分子按照摩尔比1: 50~100000在有机溶剂中进行反应,比例可变,反应中需加入一定量的碱,反应时间为0.1~100小时,可得到加氢反应产物。
[0022]
进一步的,所述有机溶剂为甲醇、乙醇、正丙醇、异丙醇、叔丁醇、四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环或甲苯,其优选乙醇、异丙醇和四氢呋喃,用量为5~100ml。
[0023]
进一步的,所述碱为醇钠、醇钾、氢化钠、氢化钾、氢氧化钠或氢氧化钾,其优选氢化钾和醇钾,加入量为催化剂摩尔量的10~500倍。
[0024]
进一步的,所述反应中可加入助剂,所述助剂为氮杂环卡宾或乙腈,其优选氮杂环卡宾,加入量为催化剂摩尔量的1~20倍。
[0025]
本发明具有如下有益效果:
[0026]
本发明的锰配合物催化剂,以低价锰离子为配合物的中心,替代价格昂贵、环境污染较大的ru、os、ir等贵金属,成本较低,在催化酯、醛、酮等多种羰基衍生物分子加氢反应中均取得了良好的目标产物收率(》95%)和催化效率;
[0027]
本发明的锰配合物催化剂采用一步合成法制备,制备方法简单,稳定性高,在催化羰基衍生物加氢反应中,催化剂用量少,对环境友好,具有较好的推广应用价值和工业化前景。
附图说明
[0028]
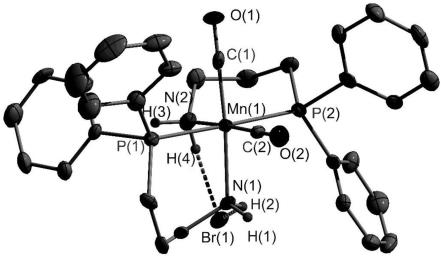
图1为实施例一中催化剂(1)的晶体结构图。
具体实施方式
[0029]
下面结合实施例对本发明的具体实施方式进行详细说明。
[0030]
一种膦胺锰配合物催化剂,所述配合物催化剂配位有四齿膦胺配体,其结构式如下:
[0031][0032]
所述配合物催化剂还可配位有双齿膦胺配体,其结构式如下:
[0033][0034]
r为苯基、五氟苯基、噻吩基、异丙基、叔丁基、硅三甲基或呋喃亚甲基;首选苯基或异丙基;
[0035]r′
、r
″
为氢、甲基、乙基、异丙基、叔丁基或苄基;首选氢或甲基;
[0036]
l5、l6为羰基、氮杂环卡宾、氢或乙腈;首选羰基或氮杂环卡宾;
[0037]
金属mn的价态为+1或+2;
[0038]
x为负一价的阴离子,其可选自不限于卤素离子、硼氢根离子、氢氧根离子、腈根离子或硫腈根离子中的任意一种;
[0039]
n的数值为0或1或2。
[0040]
金属中心mn与p、n配位点的作用可以是配位键、共价键或无相互作用;这种作用可以存在于固态,也可以存在于溶液当中;
[0041]
金属中心mn与l5、l6配位基的作用可以是配位键或共价键;这种作用可以存在于固态,也可以存在于溶液当中。
[0042]
上述金属锰配合物催化剂的制备在绝氧绝水的条件下采用标准schlenk操作技术完成,方法如下:
[0043]
在室温到回流的温度范围内,将配体l1(或l2、l3、l4)与金属锰化合物按照1:0.5~4的比例在有机溶剂中反应2~100小时,经过滤、洗涤、减压干燥得到配合物ii,有机溶剂可以是甲苯、四氢呋喃、1,4-二氧六环、乙醇、异丙醇、二氯甲烷、乙醚等,优选甲苯或四氢呋喃。
[0044][0045]
本发明中,上述金属锰配合物催化剂用于催化羰基衍生物分子加氢的活性评价过程采用高压反应釜装置完成,方法如下:
[0046]
在室温到160℃的温度范围内,起始氢气压力为10~100bar,金属配合物与底物分子取摩尔比1:50~100000(比例可变),在有机溶剂中进行反应,反应时间为0.1~100小时,可以得到加氢反应产物;
[0047]
有机溶剂可以是甲醇、乙醇、正丙醇、异丙醇、叔丁醇、四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环、甲苯等,优选乙醇、异丙醇和四氢呋喃;
[0048]
反应中需加入一定量的碱如醇钠、醇钾、氢化钠、氢化钾、氢氧化钠、氢氧化钾等,优选氢化钾和醇钾;
[0049]
反应中可以加入一定量的助剂如氮杂环卡宾、乙腈等,优选氮杂环卡宾;
[0050]
底物转化率和产物收率采用配备有氢离子火焰检测器的气相色谱仪或采用液体核磁氢谱(1h nmr)进行检测分析。
[0051]
实施例一、l1mn(co)2br(1)催化剂的制备
[0052]
惰性气氛下,称取0.14g mn(co)5br(0.5mmol)和0.243g l1(r=ph,r
′
= r
″
=h)配体(1.0mmol)于含有约40ml甲苯的schlenk瓶(100ml)中,密闭加热至80℃。反应24h后,过滤收集并用正己烷洗涤生成的黄色沉淀,减压干燥,产量约0.29g(产率86%)。
[0053]
31
p{1h}nmr(202mhz,cd2cl2,298k,ppm):δ=56.89(br).
[0054]
hrms(esi)calcd for mnc
32h36
n2p2o
2+
:597.1633,found 597.16230.
[0055][0056]
实施例二、l2mn(co)2br(2)催化剂的制备
[0057]
惰性气氛下,称取0.14g mn(co)5br(0.5mmol)和0.435g l2(r=ph,r
′
= r
″
=h)配体(1.5mmol)于含有约50ml甲苯的schlenk瓶(100ml)中,密闭加热至100℃。反应48h后,过滤收集并用正己烷洗涤生成的淡黄色沉淀,减压干燥,产量约0.35g(产率92%)。
[0058]
31
p{1h}nmr(202mhz,cd2cl2,298k,ppm):δ=59.53(br).
[0059]
hrms(esi)calcd for mnc
40h36
n2p2o
2+
:693.1633,found 693.1636.
[0060][0061]
实施例三、l3mn(co)2br(3)催化剂的制备
[0062]
惰性气氛下,称取0.14g mn(co)5br(0.5mmol)和0.39g l3(r=ph,r
′
=h) 配体(0.65mmol)于含有约40ml甲苯的schlenk瓶(100ml)中,密闭加热至80℃。反应12h后,过滤收集并用正己烷洗涤生成的黄色沉淀,减压干燥,产量约0.36g(产率91%)。
[0063]
31
p{1h}nmr(202mhz,cd2cl2,298k,ppm):δ=60.47(br).
[0064]
hrms(esi)calcd for mnc
42h38
n2p2o
2+
:719.1789,found 719.1823.
[0065][0066]
实施例四、l4mn(co)2br(4)催化剂的制备
[0067]
惰性气氛下,称取0.14g mn(co)5br(0.5mmol)和0.43g l4(r=ph,r
′
=h) 配体(0.65mmol)于含有约40ml甲苯的schlenk瓶(100ml)中,密闭加热至80℃。反应12h后,过滤收集并用正己烷洗涤生成的淡黄色沉淀,减压干燥,产量约0.40g(产率94%)。
[0068]
31
p{1h}nmr(202mhz,cd2cl2,298k,ppm):δ=62.14(br).
[0069]
hrms(esi)calcd for mnc
46h44
n2p2o
2+
:773.2259,found 773.2230.
[0070][0071]
实施例五至实施例二十二为催化剂1-4催化芳香酮加氢的方法实施例五
[0072]
在氩气氛手套箱中,称取4.1mg催化剂1、72.0mg叔丁醇钾、0.71g苯乙酮(底物酮:叔丁醇钾:催化剂=1000:100:1(摩尔比))、6ml异丙醇于50 ml反应釜中后,组装好釜体并转移出手套箱。然后,用冰水冷却釜体至 5℃,用氢气(10bar)置换釜内氩气3次后充氢气至20bar。将釜体置于加热装置中加热至100℃并在该温度下维持4h。反应完成后,快速将釜体温度降至5℃并排去釜中剩余的氢气,反应液用1cm硅胶短柱过滤后,用气相色谱 (gc)进行
分析(kb-wax色谱柱30m
×
0.32mm
×
0.33μm),1-苯乙醇收率为87%。
[0073]
实施例六
[0074]
同实施例五实验步骤,催化剂变换为2(4.6mg),得到相应产物1-苯乙醇收率为85%。
[0075]
实施例七
[0076]
同实施例五实验步骤,催化剂变换为3(4.8mg),得到相应产物1-苯乙醇收率为99%。
[0077]
实施例八
[0078]
同实施例五实验步骤,催化剂变换为4(5.1mg),得到相应产物1-苯乙醇收率为96%。
[0079]
实施例九
[0080]
同实施例七实验步骤,叔丁醇钾用量改为144.0mg(底物酮:叔丁醇钾:催化剂=1000:200:1(摩尔比)),反应时间缩短为2h,得到相应产物1-苯乙醇收率为99%。
[0081]
实施例十
[0082]
同实施例七实验步骤,反应温度改为60℃,得到相应产物1-苯乙醇收率为74%。
[0083]
实施例十一
[0084]
同实施例七实验步骤,初始氢气压力改为10bar,得到相应产物1-苯乙醇收率为78%。
[0085]
实施例十二
[0086]
同实施例七实验步骤,溶剂变换为乙醇,得到相应产物1-苯乙醇收率为 94%。
[0087]
实施例十三
[0088]
同实施例七实验步骤,溶剂变换为甲苯,得到相应产物1-苯乙醇收率为 86%。
[0089]
实施例十四
[0090]
同实施例七实验步骤,溶剂变换为四氢呋喃,得到相应产物1-苯乙醇收率为92%。
[0091]
实施例十五
[0092]
同实施例七实验步骤,苯乙酮用量改为3.55g,叔丁醇钾用量改为144.0 mg(底物酮:叔丁醇钾:催化剂=5000:200:1(摩尔比)),异丙醇用量改为 15ml,初始氢气压力改为50bar,反应时间延长至10h,得到相应产物1-苯乙醇收率为94%。
[0093]
实施例十六
[0094]
同实施例七实验步骤,酮底物变换为对氯苯乙酮,得到相应产物1-(4-氯苯基)乙醇收率为98%。
[0095]
实施例十七
[0096]
同实施例七实验步骤,酮底物变换为4-三氟甲基苯乙酮,得到相应产物1
‑ꢀ
[4-(三氟甲基)苯基]乙醇收率为99%。
[0097]
实施例十八
[0098]
同实施例七实验步骤,酮底物变换为4-甲基苯乙酮,得到相应产物1-(4-甲基苯基)-1-乙醇收率为95%。
[0099]
实施例十九
[0100]
同实施例七实验步骤,酮底物变换为4-甲氧基苯乙酮,得到相应产物1-(4
‑ꢀ
甲氧
基苯基)-1-乙醇收率为93%。
[0101]
实施例二十
[0102]
同实施例七实验步骤,酮底物变换为对硝基苯乙酮,得到相应产物1-(4-硝基苯基)乙醇收率为43%。
[0103]
实施例二十一
[0104]
同实施例七实验步骤,酮底物变换为2-甲基苯乙酮,得到相应产物1-(2-甲基苯基)乙醇收率为72%。
[0105]
实施例二十二
[0106]
同实施例七实验步骤,酮底物变换为3-甲基苯乙酮,得到相应产物1-(3-甲基苯基)乙醇收率为92%。
[0107]
表1、实施例九至实施例二十二中锰催化剂3催化不同芳香酮加氢的综合结果
[0108][0109]
[0110]
实施例二十三至实施例二十七为催化剂1-4催化芳香酸酯加氢的方法实施例二十三
[0111]
在氩气氛手套箱中,称取6.2mg催化剂1、14.0mg甲醇钾、0.136g苯甲酸甲酯(底物酯:甲醇钾:催化剂=100:20:1(摩尔比))、6ml四氢呋喃于 50ml反应釜中后,组装好釜体并转移出手套箱。然后,用冰水冷却釜体至 5℃,用氢气(10bar)置换釜内氩气3次后充氢气至50bar。将釜体置于加热装置中加热至100℃并在该温度下维持10h。反应完成后,快速将釜体温度降至5℃并排去釜中剩余的氢气,反应液用1cm硅胶短柱过滤后,用气相色谱 (gc)进行分析(kb-wax色谱柱30m
×
0.32mm
×
0.33μm),苯甲醇收率为 91%。
[0112]
实施例二十四
[0113]
同实施例二十三实验步骤,催化剂变换为2(7.0mg),得到相应产物苯甲醇收率为96%。
[0114]
实施例二十五
[0115]
同实施例二十三实验步骤,催化剂变换为3(7.3mg),得到相应产物苯甲醇收率为87%。
[0116]
实施例二十六
[0117]
同实施例二十三实验步骤,催化剂变换为4(7.7mg),得到相应产物苯甲醇收率为82%。
[0118]
实施例二十七
[0119]
同实施例二十四实验步骤,甲醇钾用量变换为70.0mg,苯甲酸甲酯用量变换为1.36g(底物酯:甲醇钾:催化剂=1000:100:1(摩尔比)),反应温度提高至120℃,反应时间延长至16h,得到相应产物苯甲醇收率为85%。
[0120]
实施例二十八至实施例三十二为催化剂1-4催化芳香醛加氢的方法实施例二十八
[0121]
在氩气氛手套箱中,称取4.1mg催化剂1、144.0mg叔丁醇钾、0.63g苯甲醛(底物醛:叔丁醇钾:催化剂=1000:200:1(摩尔比))、6ml异丙醇于 50ml反应釜中后,组装好釜体并转移出手套箱。然后,用冰水冷却釜体至 5℃,用氢气(10bar)置换釜内氩气3次后充氢气至20bar。将釜体置于加热装置中加热至100℃并在该温度下维持2h。反应完成后,快速将釜体温度降至5℃并排去釜中剩余的氢气,反应液用1cm硅胶短柱过滤后,用气相色谱 (gc)进行分析(kb-wax色谱柱30m
×
0.32mm
×
0.33μm),苯甲醇收率为 96%。
[0122]
实施例二十九
[0123]
同实施例二十八实验步骤,催化剂变换为2(4.6mg),得到相应产物苯甲醇收率为98%。
[0124]
实施例三十
[0125]
同实施例二十八实验步骤,催化剂变换为3(4.8mg),得到相应产物苯甲醇收率为99%。
[0126]
实施例三十一
[0127]
同实施例二十八实验步骤,催化剂变换为4(5.1mg),得到相应产物苯甲醇收率为99%。
[0128]
实施例三十二
[0129]
同实施例二十八实验步骤,苯甲醛用量改为3.15g(底物醛:叔丁醇钾:催化剂=
5000:200:1(摩尔比)),异丙醇用量改为15ml,初始氢气压力改为 50bar,反应时间延长至10h,得到相应产物苯甲醇收率为96%。
[0130]
本发明的锰配合物催化剂采用一步合成法制备,制备方法简单,稳定性高。以低价锰离子为配合物的中心,替代价格昂贵的ru、os、ir等贵金属,成本较低,对环境友好。该催化剂金属中心配位有一个四齿膦胺配体或两个对称的双齿膦胺配体,在催化酯、醛、酮等多种羰基衍生物分子加氢反应中均取得了良好的目标产物收率(》95%)和催化效率,值得推广使用。
[0131]
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明的范围内。本发明要求保护范围由所附的权利要求书及其等同物界定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1