一种低钙磷比的微弧氧化电解液的制作方法
本发明属于镁合金微弧氧化领域,涉及一种低钙磷比的微弧氧化电解液。
背景技术:
作为生物可降解性植入体材料,镁合金具有以下优点:密度及弹性模量与骨组织较为接近,能有效的减少应力遮蔽效应,从而促进骨组织生长;具有良好的物理性能及机械性能,适用于骨组织修复及替代;镁是人体所含的基本元素,对人体无毒性,其溶解过程不产生任何副作用;骨骼系统中的镁有利于骨骼生长,提高骨强度。然而理想的生物可降解性镁合金还需要达到较好的机械强度和完整性,较高的植入初期耐蚀性,均匀可控的降解速率及降解产物含量不超过人体能吸收的范围等方面的要求。
选择锶作为镁合金植入体的合金元素具有潜在的可行性。锶元素与钙元素同处于元素周期表中的第二主族且位于钙元素的下方,锶元素能够促进植入体的生成。锶在人体中属于微量金属元素且其总量的99%分布于骨组织中。含锶羟基磷灰石的生成有利于骨修复。锶元素能够促进骨植入体周围细胞的生长,繁殖和修复,从而提高骨诱导和骨形成能力。另外,研究发现含2%质量分数的sr的镁合金具有较高的力学性能和较低的耐蚀性能。综上所述,mg-2sr合金可以作为一种潜在的生物相容性镁基合金用于后续的实验。然而,镁在人体内的降解速度过快,无法在新骨组织长成之前维持支撑作用,降解产生的氢气沉积在植入体周围,阻碍了伤口愈合,同时也提高了植入体周围的ph值,甚至发生碱中毒。提纯镁,合金化及表面改性可以作为增强耐蚀性的三种不同解决方案。实验采用锶元素合金化制备性能优良的mg-2sr合金,随后利用微弧氧化技术在其表面制备功能性膜层以实现合金化和表面改性的综合利用。微弧氧化(microarcoxidation,mao)又称微等离子体氧化,是利用电化学方法在al,mg,ti,zr,nb,ta等金属表面施加电压至产生火花放电现象,在热化学、等离子体化学和电化学的共同作用下,经历熔融、喷发、结晶、高温相变,最终在基体表面熔覆,烧结形成陶瓷层。微弧氧化膜层具有高硬度,良好的耐磨性,较好的耐蚀性及热稳定性。陶瓷膜层与金属基体之间呈冶金结合从而具有较强的结合力。
合金基体,电参数及电解液配方综合影响了微弧氧化膜层的性能。其中,来自电解液配方中的组分将伴随微弧氧化反应进入到膜层中从而影响膜层的相组成,微观结构,耐蚀性能,结合力及生物降解性能等。所以,相对于既定的基体及电参数来说,电解液配方对于膜层最终的性能具有决定性的作用。
技术实现要素:
为弥补现有传统电解液的不足,本发明提供一种添加碳酸钙的微弧氧化电解液体系。
本发明是通过以下方式实现的:
一种低钙磷比的微弧氧化电解液,ca/p≤1/2。
优选的,所述电解液的ca/p为1/7~1/2,优选的ca/p为1/6~1/2,进一步优选的ca/p为1/5-1/3。
优选的,电解液所用的钙源为碳酸钙,碳酸钙的浓度为0.2-4g/l;优选的,碳酸钙的浓度为0.3-3g/l;进一步优选的,碳酸钙的浓度为0.5-1g/l;最优选的,碳酸钙的浓度为0.600g/l。
本发明的电解液还包括以下一种或多种组分及质量浓度:(na2po3)6的浓度为1-30g/l,koh的浓度为2-10g/l,nh4hf2的浓度为5-10g/l,c3h8o3的浓度为2~10ml/l,n(ch2ch2oh)3的浓度为2-10ml/l,h2o2的浓度为1-13ml/l。
本发明优化的一种或多种组分及质量浓度为:(na2po3)6的浓度为1-20g/l,koh的浓度为4-6g/l,nh4hf2的浓度为6-8g/l,c3h8o3的浓度为6~8ml/l,n(ch2ch2oh)3的浓度为4-6ml/l,h2o2的浓度为4-10ml/l。
本发明进一步优化的一种或多种组分及浓度为:(na2po3)6的浓度为1.836g/l或2.448g/l或3.060g/l,koh的浓度为5g/l,nh4hf2的浓度为7g/l,c3h8o3的浓度为5ml/l,n(ch2ch2oh)3的浓度为5ml/l,h2o2的浓度为7ml/l。
优选的,所述电解液的ca/p为1/2、1/3、1/4或1/5。
本发明还提供了一种镁合金表面含锶羟基磷灰石微弧氧化涂层的制备方法,在镁合金基体上采用微弧氧化法制备涂层;
所述微弧氧化法采用的电解液为任一上述的电解液;
所述镁合金基体为mg-2sr。
优选的,所述的微弧氧化处理具体步骤为:将预处理的镁合金基体晾干置于电解液中作为正极,不锈钢槽作为负极,通循环冷却水保持电解液温度控制在50℃以下,采用微弧氧化电源供电,电源频率500~600hz,正占空比20%~40%,负占空比10%~30%,添加负向电压20-60v,在正向电压400v~500v的恒压下通电反应5~20min;优选的,微弧氧化电源参数为电源频率550hz,正占空比30%,负占空比20%,添加负向电压40v,在正向电压450v的恒压下通电反应10min,所述的微弧氧化电源为双向脉冲电源、单向脉冲电源或直流电源。
本发明还提供了一种低钙高磷添加碳酸钙的电解液在提高镁合金微弧氧化涂层耐腐蚀性和/或生物学性能(生物相容性或生物活性)上的应用。本发明通过对镁锶合金的微弧氧化膜成膜规律的系统研究发现:与其它基体材料不同,对镁锶合金而言,添加含碳酸钙的、低钙高磷电解液不仅可使其微弧氧化膜具有较优的生物学性能(生物相容性或生物活性),而且可使其微弧氧化膜的耐腐蚀性较现有高钙低磷电解液提高2个数量级以上。
本发明的有益效果:
1)本发明制备的微弧氧化膜层都具有典型的微孔结构并且没有发现较为明显的微裂纹,微弧氧化膜层表面的颗粒状腐蚀产物中钙磷相含量较高,生物学性能(生物相容性或生物活性)良好。微弧氧化膜层较基体具有较好的耐蚀性,较现有高钙低磷电解液提高2个数量级以上。
2)本发明分别选择葡萄糖酸钙,乳酸钙和碳酸钙为钙源,六偏磷酸钠为磷源的电解液体系(钙磷比确定为1:2),采用恒定正向电压和负向电压通过微弧氧化技术在自行铸造的mg-2sr合金表面制备钙磷膜层xrd和ft-ir红外测试结果显示模拟体液浸泡前的微弧氧化膜层中所含的相主要为mg,mgo,sr2mg17,mgf2,cao,caf2,ca3(po4)2(tcp)等,另外在浸泡后膜层中还检测到了ca2p2o7,mg(oh)2,ha,sr-ha等新相的存在。由葡萄糖酸钙为钙源制备的微弧氧化膜层厚度较小,由乳酸钙,碳酸钙为钙源制备的微弧氧化膜层厚度较大且较为接近。三种微弧氧化膜层都具有典型的微孔结构并且没有发现较为明显的微裂纹。模拟体液浸泡试验表明由碳酸钙为钙源制备的微弧氧化膜层表面的颗粒状腐蚀产物中钙磷相含量较高,生物学性能(生物相容性或生物活性)良好。微弧氧化膜层较基体具有较好的耐蚀性,其中由乳酸钙,碳酸钙为钙源制备的微弧氧化膜b和c的失重率也较低。
3)本发明采用恒定正向电压和负向电压通过微弧氧化技术在自行铸造的mg-2sr合金表面制备钙磷膜层xrd和ft-ir红外测试结果显示模拟体液浸泡前的微弧氧化膜层中所含的相主要为mg,mgo,sr2mg17,mgf2,cao,caf2,ca3(po4)2(tcp)等,另外在浸泡后膜层中还检测到了ca2p2o7,mg(oh)2,ha,sr-ha等新相的存在。其中,以乳酸钙为钙源的膜层中随着钙磷比的降低,膜层表面变得更加平整,微孔尺寸变得更加细小均匀,微孔数量也明显增加。碳酸钙为钙源的膜层中钙磷比对膜层表面形貌影响较小。模拟体液浸泡试验表明两种钙源下,钙磷比为1:4时制备的微弧氧化膜层表面的颗粒状腐蚀产物中钙磷相含量较高,生物学性能(生物相容性或生物活性)良好且具有较好的耐蚀性,失重率及降解速率都较低。
附图说明
图1不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层的xrd图谱;
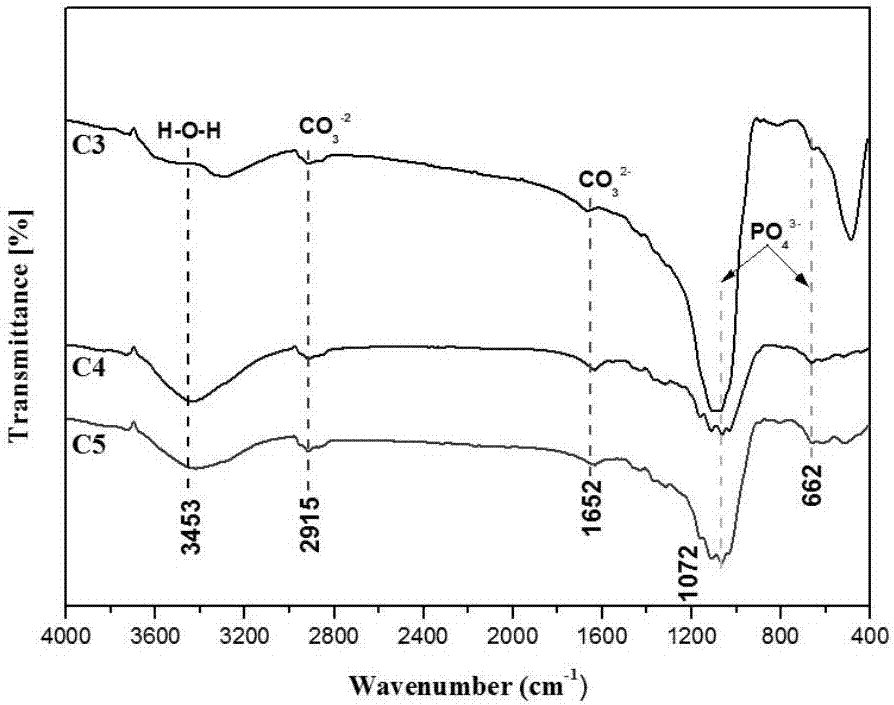
图2不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层的ft-ir图谱;
图3碳酸钙为钙源的电解液制备的微弧氧化膜层形貌和元素分析:(a)c3,(b)c4,(c)c5;
图4c4膜层表面形貌及元素分布图;
图5含碳酸钙的电解液制备的微弧氧化膜层截面形貌和元素分析:(a)c3,(b)c4,(c)c5;
图6不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层的结合力;
图73种不同钙磷比的含碳酸钙电解液体系中微弧氧化膜层在sbf中的tafel极化曲线;
图8不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层在sbf中浸泡21天后的xrd图谱;
图9不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层在sbf中浸泡14天后的ft-ir图;
图10碳酸钙为钙源的微弧氧化膜层sbf浸泡21天后的表面形貌和元素原子百分比:c3(a1,a2),c4(b1,b2),c5(c1,c2);
图11不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层在sbf中浸泡不同天数后的失重情况;
图12不同钙磷比含碳酸钙电解液制备的微弧氧化膜层sbf浸泡28天的降解速率;
图13不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层sbf浸泡过程中的ph值随时间的变化曲线。
具体实施方式
下面结合实施例进一步说明:
实施例1
1.本实验中制备的微弧氧化膜层涉及到了三种不同的电解液体系,分别是:以碳酸钙为钙源,钙磷摩尔比设定为1:3、1:4和1:5,分别定义为c3、c4和c5。本实验系统地对比研究了上述三种不同钙源电解液体系下制备的微弧氧化膜层的性能,给出了上述碳酸钙钙源下的优化钙磷摩尔比。
2.实验材料与方法
2.1基体与膜层制备
实验所用基体为自行铸造的mg-2sr合金,具体步骤为:使用气氛保护(sf6:co2=1:200)电阻炉熔解纯镁(99.99%)和镁锶中间合金(mg-21sr(99.99%))。所有原材料和铸造所用工具均需要预热至250℃。炉温升至500℃时,放入预热后的纯镁锭。炉温为700℃时纯镁完全熔解并保温20min。调整炉温至710℃时加入理论量的镁锶中间合金(mg-21sr(99.99%)),保温20min。充分搅拌熔体并浇注到预热好的石墨模具中。随后对浇注完成的mg-2sr合金进行400℃下均匀化退火16个小时。再利用线切割设备将铸体切成规则的8mm×10mm×10mm立方体小块,随后经粗360#、粗600#、细600#、细1000#砂纸打磨砂纸进行机械打磨。最后用丙酮,去离子水,无水乙醇清洗并烘干待用。微弧氧化实验所用的钙源为碳酸钙caco3,钙离子摩尔浓度均为0.006mol/l,质量浓度为0.600g/l;实验所用磷源为(na2po3)6,浓度分别为1.836g/l、2.448g/l和3.060g/l;剩余电解液组分及浓度为koh,5g/l;nh4hf2,7g/l;c3h8o3和n(ch2ch2oh)3浓度均为5ml/l;h2o2浓度为7ml/l。钙磷比分别为1:3/1:4和1:5,对应的电解液体系定义为c3、c4和c5。实验所用药品均为分析纯。微弧氧化实验过程中,镁合金试样作为正极,不锈钢槽作为负极,通循环冷却水保持电解液温度在50℃以下,采用微弧氧化电源供电,具体电参数如表1.
表1微弧氧化电参数
2.2.膜层检测
1,膜层相组成分析:采用x射线衍射仪(brukerd8advanced,germanyandshmadzuxrd-6100)测试分析微弧氧化膜层的相组成。测试条件为:cu靶(cu-kα),管电压40kv,管电流30ma,扫描范围10~90°,扫描速度为4°/min,计数器的样间隔为0.02。采用德国brook公司tensor37型号傅里叶变换红外光谱仪检测分子结构及官能团的变化,红外透射光谱波数范围为4000cm-1~400cm-1,分辨率为4cm-1,扫描时间为16s。
2,膜层微观形貌及成分分析(sem,eds):采用日立(hitachi)公司的s-3400n测试分析浸泡前后膜层的表面形貌,用日本电子(jeol)公司的jsm-6380la扫描电镜装配jed-2300型能谱分析仪(eds)对膜层进行成分分析。对不导电的试样,测试前进行喷金处理。喷金设备采用北京中科科仪技术发展有限责任公司研制的kykysbc-12型离子溅射仪,所用靶材为金靶,电流20ma,喷金时间120s。
3,膜层厚度测量:采用型号为minitest600bfn2的厚度测试仪对膜层的厚度进行测量,选择测试非磁性金属基体上的非导电覆盖层的厚度,最终膜厚值为5次测试值的平均值。
4,划痕测试:划痕法测试采用金刚石划针在恒定或连续增加的正压力、一定的速度下刻划膜层表面,直至发生破坏,以其对应的临界载荷作为膜层与镁合金基体的结合强度。划痕实验采用中国科学院兰州化学物理研究所研制的ws-2005型划痕仪,金刚石锥角120°,曲率半径0.2mm。最大加载力为30n,划痕速度为30n/min,工作台移动速度为3mm/min。
2.3降解及生物活性测试
采用体外模拟体液浸泡试验对微弧氧化膜层的生物活性进行表征。1.0sbf溶液所含离子浓度与人体血浆所含离子浓度类似,由nacl,nahco3,kcl,k2hpo4·3h2o,mgcl2·6h2o,1.0mol/lhcl,cacl2和na2so4配制而成,最后由(ch2oh)3cnh2和盐酸调节溶液ph为7.2-7.4,整个配置过程要求溶液温度保持在36.5±0.5℃。然后将待浸泡试样放入装有模拟体液的塑料瓶中,静置于36.5℃的恒温水浴槽中。试样浸泡过程中,模拟体液每隔一天更换一次并保持无色无沉淀。试样浸泡1天,7天,14天,21天和28天后,分别取出样品,依次用去离子水和丙酮清洗,然后用鼓风式干燥箱充分干燥。
2.4电化学性能测试
电化学性能测试:采用美国princetonappliedresearch公司生产的princetonparstat2273电化学工作站测试膜层的动电位极化曲线,并与基体进行对比。电化学测试采用标准三电极体系,以模拟体液为腐蚀介质,待测试样(面积1.00cm2)为工作电极,铂片(面积1.00cm2)作辅助电极,ag/agcl电极为参比电极,扫描电压范围为-2000mv~-1300mv,扫描速度设定为1mv/s。
3.实验结果
3.1膜层的组织形貌
3.1.1膜层的物相
图1为不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层的xrd图谱。可以看出,碳酸钙钙源下不同钙磷比的电解液体系制备的微弧氧化膜层的物相基本一致,均由mg,mgo,sr2mg17,mgf2,cao,caf2,ca3(po4)2(tcp)等相组成。其中,ca3(po4)2(tcp)相的存在表明基体外部电解液中的ca,p元素已成功地通过微弧氧化过程进入到膜层中。另外,tcp相具有良好的生物学性能(生物相容性或生物活性)和可降解性,被广泛应用于临床领域。植入体内后,tcp相还会与体液反应生成ha。具体的,c3曲线中的峰较为密集且峰值相对较弱,基体相mg,mgo的峰强相对其他两条曲线更为微弱,表明c3膜层的膜厚较大,因而x射线很难穿透膜层进入基体内部,c4曲线中37°处的cao的峰值很强,表明该相在膜层c4中结晶良好,c5曲线中的各物相的峰强较强尤其是70°处基体mg相的峰强很高,表明c5膜层厚度可能较小,x射线很容易穿透膜层进入基体内部。综上所述,膜层c4,c5中的各物相结晶程度较大,而膜层c3中有非晶相的存在导致其峰强较低。
3.1.2膜层的ft-ir功能基团分析
图2为不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层的ft-ir图谱,由图2可知,由碳酸钙为钙源制备的微弧氧化膜层中出现了2915cm-1和1652cm-1处co32-的劈裂特征峰,另外还出现了3453cm-1处的o-h伸缩振动峰,1072cm-1和652cm-1处的峰对应的是po43-,hpo42-或p2o74-。此外,低于500cm-1以下的峰被认为是m-o或者m-f(m=mg,ca,sr)。具体的,碳酸钙与六偏磷酸钠比例为1:4和1:5的膜层c4和c5的ft-ir曲线形状类似,特征峰均较为宽而平缓,当碳酸钙与六偏磷酸钠比例为1:3时,c3膜层的ft-ir曲线中的特征峰较为尖锐,尤其体现在1072cm-1处的磷酸根基团的特征峰,此外,c3曲线中还在485cm-1处出现了一个较为尖锐的特征峰,这可能是m-o或者m-f(m=mg,ca,sr)。综上所述,电解液体系中钙磷比的变化会在不同程度上影响到ft-ir曲线中特征峰的形状,同一钙源下制备的微弧氧化膜层中所检测到的功能基团相同。
3.1.3膜层的表面形貌及成分分析
图3为以碳酸钙为钙源的电解液制备的微弧氧化膜层形貌和元素分析。其中图3(a)的膜层c3表面的微孔零星分布于结节处周围,微孔尺寸范围在10-20μm之间,部分较大的孔洞内部沉积了白色絮状物质,膜层表面有局部的结节凸起,结节处出现了细长的微裂纹。再由图3(b)可知,随着钙磷比降低至1:4,膜层c4表面的微孔数量明显增加,微孔尺寸更加细小均匀,在5-10μm之间。另外,膜层表面更加平整,微孔内部没有发现多余的沉积物出现,膜层c4中的微裂纹较为稀少。随着钙磷比的进一步降低,如图3(c),膜层表面出现了局部分布的更加细小的微孔,尺寸仅有2μm左右,同时也有尺寸较大,约为15μm的微孔。部分较大微孔内部堆满了颗粒状沉积物,整个膜层表面较为粗糙,没有发现细长的微裂纹的存在。综上所述,以碳酸钙为钙源的膜层中随着钙磷比的降低,膜层表面的微孔先变得更加细小均匀,随后出现了较大的微孔尺寸,同时膜层表面先变得更加平整光滑随后粗糙度有所增加。再由三种膜层表面的两处不同位置的元素分析结果可知,所有膜层中均含有mg,o,ca,p,f,c等元素,这些元素中除了基体元素mg,其他元素均来自电解液体系,这表明电解液中的成分成功地通过微弧氧化反应进入到膜层内部。由于检测误差及膜层中sr元素含量较低从而未能检测出基体的sr元素。
3.1.4膜层的面扫描及成分分析
根据前述实验结果的综合分析,本部分选择对试样c4膜层表面进行了面扫描,根据图4,c4膜层表面的微孔更加均匀细小并分布于膜层的整个表面,没有出现细长状的较深的孔洞。膜层同样由mg,sr,o,ca,p,f等元素组成,其中mg,o,f,p元素主要分布在孔洞以外的凸起部位,ca元素和sr元素弥散分布于整个区域,明显地,ca元素比sr元素的分布更加密集,即在膜层表面的含量更高,这与图3的分析结果相一致。其次,ca,p,o三种元素同时出现在凸起结构及其周围,孔洞处三种元素的分布较为稀少但能够被检测到,表明这些位置分布着钙磷酸盐因而具有较高的生物活性,在模拟体液中有较强的诱导生成羟基磷灰石的能力。
3.1.5膜层的截面形貌及膜层结合力
图5为以碳酸钙为钙源的电解液制备的微弧氧化膜层截面形貌和元素分析。由图可知,三种钙磷比下制备的微弧氧化膜层与基体和环氧树脂之间的结合情况各有不同,膜层的厚度差别也较大。具体的,钙磷比为1:3的膜层c3厚度最小,约为40μm,该膜层致密层与基体连接十分紧密,致密层与疏松层过渡良好,其厚度稍大于疏松层的厚度,疏松层与环氧树脂之间存在明显的界限,剥落现象较为明显;钙磷比为1:4的膜层c4厚度约为70μm,膜层质量优良,致密层与基体之间连接十分紧密,没有出现剥落现象,疏松层区域出现了裂纹和微孔,这可能是由于微弧氧化过程中出现较大的放电火花所致,疏松层的厚度明显大于致密层厚度。钙磷比为1:5的膜层c5厚度为50μm左右,其致密层较薄且与基体连接紧密,致密层和疏松层过渡较差,疏松层区域较大且较为松散,内部出现了孔洞和剥落现象,其中存在一条横穿的微裂纹直至环氧树脂区域,表明该膜层的疏松层质量较差,这可能是由于微弧氧化过程中导电率较大从而产生过大的放电火花导致的。其次,疏松层与环氧树脂的连接区域出现了明显的裂痕和缝隙。由膜层截面线扫描结果可知,其元素种类依然是mg,o,ca,p,f,c,sr等,三种膜层的横截面元素分布图十分类似,沿着膜层内部深度方向上,mg元素含量逐渐上升,骤增处为致密层和基体的结合区域。另外,在膜层内部检测到了o,ca,p,f等元素,说明这些元素是通过微弧氧化反应进入到膜层中,并对改变膜层的组织与结构起到一定的作用。
图6为以碳酸钙为钙源的电解液制备的微弧氧化膜层的结合力示意图。由图可知,随着钙磷比的降低,膜层与基体的结合力呈现上升的趋势,结合力数值在20-30n之间,具体的,c3,c4,c5膜层的结合力分别为21.2n,25.4n,28.1n,这与上述的膜层截面形貌表现出的规律相对比较吻合。
3.2膜层的耐蚀性能
图7为不同电解液体系中微弧氧化膜层在sbf中的tafel极化曲线,根据tafel极化曲线外推法得出表2中各曲线对应的腐蚀电流icorr及腐蚀电压ecorr。如前所述,这些数据可以定性地比较不同材料的腐蚀倾向及腐蚀速率。由图7和表2中的数据可知,以碳酸钙为钙源制备的微弧氧化膜层c3,c4,c5的腐蚀电流较为相近,均比基体降低了2个数量级,代表三种膜层的腐蚀速率较低,耐蚀性较好,腐蚀电压相对于基体正向移动了0.234-0.508v。具体的,钙磷比为1:3的膜层c3腐蚀电压正向移动了0.508v,腐蚀电流最小,为1.889*10-7a/cm2,这说明c3膜层最难被腐蚀,腐蚀速率也最低。钙磷比为1:4的膜层c4的腐蚀电流正向移动了0.234v,而腐蚀电流仍然很低,仅有2.002*10-7a/cm2,这说明膜层c4较易被腐蚀,但实际腐蚀速率却较低,这可能是因为膜层c4的疏松层较为松散,腐蚀性离子很容易进入疏松层,但致密层的结构较为均匀致密且与基体结合良好,因而最终的腐蚀速率仍然较低。钙磷比为1:5的膜层c5的腐蚀电压负向移动了0.347v,而腐蚀电流仅有2.113*10-7a/cm2,这与图5所描述的膜层横截面现象基本一致。
表2不同电解液体系中微弧氧化膜层电化学测试数据
3.3膜层的生物活性及体外降解行为
3.3.1sbf浸泡后膜层相组成及ft-ir功能基团分析
图8为不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层在sbf中浸泡21天后的xrd图谱,和图1相比,除了溶解度较高的tcp相,浸泡之前所能检测的物相基本都还存在,另外,在浸泡21天后的膜层表面检测到了ca2p2o7(ccp),mg(oh)2,ha和ca10-xsrx(po4)6(oh)2(sr-ha)等相的存在,其中ca10-xsrx(po4)6(oh)2是元素sr取代了羟基磷灰石中元素ca的位置而形成的一种新的生物相容性磷灰石,该相可以促进骨植入体周围的细胞的生长、繁殖和修复,ccp相同样也是一种具有良好生物学性能(生物相容性或生物活性)的材料。ha被认为是用于硬组织修复最合适的陶瓷材料,它没有任何毒性,且可以直接与骨细胞结合。图8中的三条曲线的差别十分明显,曲线c3的峰十分尖锐而曲线c4,c5的峰相对较低。其中,曲线c3中基体相、ccp、ha、sr-ha的峰最为尖锐,说明这些物相结晶度很高,另外c3试样浸泡后的腐蚀产物可能较少,堆积层较薄,因而x射线很容易穿透腐蚀产物达到基体。曲线c4和c5的峰较低而宽,表明膜层c4和c5浸泡后的表面出现了非晶相,其中c5曲线在63°处出现了ha的峰且较为尖锐,其他两条曲线中ha相的峰十分微弱。
图9为不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层在sbf中浸泡14天后的ft-ir图谱,和图2相比,浸泡后的3种膜层的ft-ir图谱发生了较大的变化,而浸泡后的3种膜层的ft-ir图谱十分相近,并同时在3700cm-1处出现了尖锐的oh-1的伸缩振动峰,表明浸泡后的膜层中出现了mg(oh)2,这与浸泡后的xrd检测出mg(oh)2的结果相一致。其次,3种膜层浸泡后都出现了自由水在3420cm-1处较宽的拉伸吸收峰,此外,由乳酸钙制备的膜层浸泡后的ft-ir图谱中也检测到了1439cm-1处的co32-的v3反对称伸缩峰,和图2相比,po43-,hpo42-或p2o74-等基团的峰形变化也相对较少,由原先的1072cm-1和662cm-1处的峰形变为1056cm-1和570cm-1处的峰形,另外曲线c5中也出现了870cm-1处磷酸根基团的峰。
3.3.2膜层的表面形貌及成分分析
图10为以碳酸钙为钙源的微弧氧化膜层sbf浸泡21天后的表面形貌和元素成分分析,具体的,由图10(a1)(a2)可知,膜层c3在sbf中浸泡21天后部分保留了多孔结构,其余部分腐蚀较为严重,整个表面出现凹凸不平的现象,在放大图中发现了密集分布的腐蚀裂纹,膜层表面和微裂纹周围分布着层片状的腐蚀产物,孔洞处沉积的腐蚀产物较少。由图10(b1)(b2)可知,膜层c4表面较为平整,出现了少量的微裂纹,微孔深处和周围堆积了颗粒状的腐蚀产物,再由放大图可知,腐蚀产物中出现了纳米棒和纳米线状的物质,并沿腐蚀裂纹分布,中间夹杂着层片状的腐蚀产物。由图10(c1)(c2)可知,膜层保留着部分完整的微孔结构,部分区域出现了层片状的腐蚀产物,再由放大图可知,层片状的腐蚀产物周围出现了大量的纳米线状的物质,宛如一朵菊花,该部分腐蚀产物生长在腐蚀微裂纹处,表明该处具有较大的能量易于形核。元素分析结果给出了三种膜层腐蚀产物处的元素原子种类和比例,均包含了c,o,f,mg,ca,p,sr等元素。具体的,膜层c3中的ca,p,o三种元素的原子数百分比较高,分别为18%,15%和47%,说明该腐蚀产物中含有较多的钙磷化合物,生物学性能(生物相容性或生物活性)良好。膜层c4的腐蚀产物中o和mg元素含量较高,表明腐蚀产物中含有较多的mgo,其次f,p,ca元素含量接近,约10%。膜层c5的腐蚀产物中,ca,p,o三种元素的原子数百分比较高,分别为22%,18%和42%,f,mg元素含量都较低,表明膜层c5表面的腐蚀产物中含有大量的钙磷化合物,大大地提高了膜层的生物活性。
3.3.3试样失重及ph值变化
试样的体外降解行为可以通过失重率、降解速率和ph曲线来评估。试样最终表现出的失重率应是膜层降解和磷灰石相等腐蚀产物生成两者综合作用的结果。根据图11,三种膜层在浸泡前7天的腐蚀降解速率都较低,浸泡14天后,膜层c3的失重率最大,约为12%,而膜层c4的失重率仅有3.2%,膜层c5的失重率为8.7%。浸泡21天结束后,膜层c4的失重率最低,约为13%,膜层c3和c5的失重率约为15%和17%。综上所述,钙磷比为1:4的膜层b4和c4具有较低的失重率和较强的耐蚀性,其有利于提高植入体在体内的稳定性从而更好地完成支撑服役任务,这与电化学测试结果相一致。
图12为不同钙磷比的含碳酸钙的电解液制备的微弧氧化膜层sbf浸泡21天的降解速率,其中c3,c4,c5的降解速率分别为0.24015,0.17684,0.19481(mg/(cm2·d))。可以看出,钙磷比为1:4的膜层c4的失重率和降解速率最低,表明膜层c4的耐蚀性较好,前期的降解也较慢,这有利于保障植入体维持体内的支撑任务。
图13为不同钙磷比的含碳酸钙电解液制备的微弧氧化膜层sbf浸泡过程中的ph值随时间的变化曲线。原始模拟体液的ph值为7.25,在21天的浸泡周期内,模拟体液的ph值浮动较大。ph值变化的整体趋势为:膜层c3,c4,c5分别在浸泡1天,3天和5天后达到了最大值9.9,8.7和9.1,随后的浸泡周期内,ph值发生较为明显的下降,并同时在浸泡11天后降至8.4左右,浸泡11-13天,ph值发生较小程度的上浮,浸泡13-21天,ph值再次发生下降并稳定在7.4-8.2之间。具体的,在一个单位浸泡周期(2天)内,ph值随着浸泡时间的延长而上升。相同的浸泡时间内,膜层c5的ph值明显高于膜层c3和c4的ph值。其中,膜层c4的ph值变化幅度较小并且在第3天出现了最大值。
4结论
(1)浸泡前微弧氧化膜层主要含有的物相有mg,mgo,sr2mg17,mgf2,cao,caf2,ca3(po4)2(tcp)等,浸泡后的膜层中溶解度较高的tcp相消失,并检测到了ca2p2o7(ccp),mg(oh)2,ha和ca10-xsrx(po4)6(oh)2(sr-ha)等新相的存在。
(2)以碳酸钙为钙源的膜层中随着钙磷比的降低,膜层表面的微孔先变得更加细小均匀,随后出现了较大的微孔尺寸,膜层表面先变得更加平整光滑随后粗糙度有所增加。
(3)以碳酸钙为钙源制备的微弧氧化膜层的腐蚀电流较为相近,均比基体降低了2个数量级,代表三种膜层的腐蚀速率较低,耐蚀性较好。
(4)浸泡21天后的膜层的表面形貌受到了不同程度的腐蚀,基本都部分保留着原先的多孔结构,膜层c4和c5的腐蚀产物中出现了纳米棒和纳米线状的物质,并沿腐蚀裂纹分布,中间夹杂着层片状的腐蚀产物,ca,p,o原子百分比也较高,这正是提高材料生物活性所迫切需要的。
(5)钙磷比为1:4的膜层c4具有较低的失重率和较强的耐蚀性,在sbf中浸泡21天后的失重率仅有13%,前期的降解也较慢,对应于21天的降解速率在同组中最低,这有利于提高植入体在体内的稳定性从而更好地完成支撑服役任务,其次,膜层c4在浸泡过程中的ph值变化幅度相对其他膜层较小并且在第3天出现了最大值。
实施例2
本实施例中制备的微弧氧化膜层涉及到了三种含不同钙源的电解液体系,包括研究较多的葡萄糖酸钙以及研究较少的乳酸钙,碳酸钙等。上述三种钙源的钙含量分别为9%,13%和40%。有机钙源葡萄糖酸钙和乳酸钙中的钙含量明显低于无机钙源碳酸钙中的钙含量。本实验系统地对比研究了上述三种不同钙源电解液体系下制备的微弧氧化膜层的性能,表明了有机钙和无机钙制备出的微弧氧化膜层所存在的差异。
2.实验材料与方法
2.1基体与膜层制备
实验所用基体为自行铸造的mg-2sr合金,具体步骤为:使用气氛保护(sf6:co2=1:200)电阻炉熔解纯镁(99.99%)和镁锶中间合金(mg-21sr(99.99%))。所有原材料和铸造所用工具均需要预热至250℃。炉温升至500℃时,放入预热后的纯镁锭。炉温为700℃时纯镁完全熔解并保温20min。调整炉温至710℃时加入理论量的镁锶中间合金(mg-21sr(99.99%)),保温20min。充分搅拌熔体并浇注到预热好的石墨模具中。随后对浇注完成的mg-2sr合金进行400℃下均匀化退火16个小时。再利用线切割设备将铸体切成规则的8mm×10mm×10mm立方体小块,随后经粗360#、粗600#、细600#、细1000#砂纸打磨砂纸进行机械打磨。最后用丙酮,去离子水,无水乙醇清洗并烘干待用。微弧氧化实验所用的三种钙源为葡萄糖酸钙ca(c6h11o7)2﹒h2o,乳酸钙ca(c3h5o3)2﹒5h2o和碳酸钙caco3,钙离子摩尔浓度均为0.006mol/l,质量浓度则分别为2.688g/l,1.848g/l,0.600g/l和0.444g/l;剩余电解液组分及浓度为(na2po3)6,1.224g/l(0.012mol/l);koh,5g/l;nh4hf2,7g/l;c3h8o3和n(ch2ch2oh)3浓度均为5ml/l;h2o2浓度为7ml/l。三组含不同钙盐的电解液体系分别定义为a,b,c,其中钙磷比均为1:2。实验所用药品均为分析纯。微弧氧化实验过程中,镁合金试样作为正极,不锈钢槽作为负极,通循环冷却水保持电解液温度在50℃以下,采用微弧氧化电源供电,具体电参数如表3.
表3微弧氧化电参数
2.2.膜层检测
1,膜层相组成分析:采用x射线衍射仪(brukerd8advanced,germanyandshmadzuxrd-6100)测试分析微弧氧化膜层的相组成。测试条件为:cu靶(cu-kα),管电压40kv,管电流30ma,扫描范围10~90°,扫描速度为4°/min,计数器的样间隔为0.02。采用德国brook公司tensor37型号傅里叶变换红外光谱仪检测分子结构及官能团的变化,红外透射光谱波数范围为4000cm-1~400cm-1,分辨率为4cm-1,扫描时间为16s。
2,膜层微观形貌及成分分析(sem,eds):采用日立(hitachi)公司的s-3400n测试分析浸泡前后膜层的表面形貌,用日本电子(jeol)公司的jsm-6380la扫描电镜装配jed-2300型能谱分析仪(eds)对膜层进行成分分析。对不导电的试样,测试前进行喷金处理。喷金设备采用北京中科科仪技术发展有限责任公司研制的kykysbc-12型离子溅射仪,所用靶材为金靶,电流20ma,喷金时间120s。
3,膜层厚度测量:采用型号为minitest600bfn2的厚度测试仪对膜层的厚度进行测量,选择测试非磁性金属基体上的非导电覆盖层的厚度,最终膜厚值为5次测试值的平均值。
4,划痕测试:划痕法测试采用金刚石划针在恒定或连续增加的正压力、一定的速度下刻划膜层表面,直至发生破坏,以其对应的临界载荷作为膜层与镁合金基体的结合强度。划痕实验采用中国科学院兰州化学物理研究所研制的ws-2005型划痕仪,金刚石锥角120°,曲率半径0.2mm。最大加载力为30n,划痕速度为30n/min,工作台移动速度为3mm/min。
2.3降解及生物活性测试
采用体外模拟体液浸泡试验对微弧氧化膜层的生物活性进行表征。1.0sbf溶液所含离子浓度与人体血浆所含离子浓度类似,由nacl,nahco3,kcl,k2hpo4·3h2o,mgcl2·6h2o,1.0mol/lhcl,cacl2和na2so4配制而成,最后由(ch2oh)3cnh2和盐酸调节溶液ph为7.2-7.4,整个配置过程要求溶液温度保持在36.5±0.5℃。然后将待浸泡试样放入装有模拟体液的塑料瓶中,静置于36.5℃的恒温水浴槽中。试样浸泡过程中,模拟体液每隔一天更换一次并保持无色无沉淀。试样浸泡1天,7天,14天,21天和28天后,分别取出样品,依次用去离子水和丙酮清洗,然后用鼓风式干燥箱充分干燥。
2.4电化学性能测试
电化学性能测试:采用美国princetonappliedresearch公司生产的princetonparstat2273电化学工作站测试膜层的动电位极化曲线,并与基体进行对比。电化学测试采用标准三电极体系,以模拟体液为腐蚀介质,待测试样(面积1.00cm2)为工作电极,铂片(面积1.00cm2)作辅助电极,ag/agcl电极为参比电极,扫描电压范围为-2000mv~-1300mv,扫描速度设定为1mv/s。
3.结论
(1)以葡萄糖酸钙为钙源的电解液发生微弧氧化反应时,电火花较为细小且零星分布,蜂鸣声较低且持续时间较短,反应能量较低;含其他两种钙源的电解液体系在反应过程中产生大量气泡,出现大量放电火花并伴有刺耳的轰鸣声,火花持续时间较长,反应能量较高。因此膜层a的厚度较小约为25.6μm,膜层b和c厚度较大且较为接近,分别为37.8和34.0μm。
(2)浸泡前的微弧氧化膜层主要含有的物相有mg,mgo,sr2mg17,mgf2,cao,caf2,ca3(po4)2(tcp)等,浸泡后的膜层中溶解度较高的tcp相消失,并检测到了ca2p2o7(ccp),mg(oh)2,ha和ca10-xsrx(po4)6(oh)2(sr-ha)等新相的存在。
(3)由微弧氧化技术制备的膜层具有较好的耐蚀性,其中膜层b和c的腐蚀电压和腐蚀电流相对于基体都有明显地降低。任一浸泡周期内,膜层b和c的失重率相对其它浸泡组较低,说明由乳酸钙和碳酸钙为钙源的电解液制备的微弧氧化膜层降解速率较低。
(4)结合xrd,ft-ir红外谱图,edx元素分析可知,sbf浸泡过程中磷酸钙成功地转化为钙磷灰石。其中由乳酸钙为钙源制备的微弧氧化膜层b的颗粒状腐蚀产物中钙磷相含量比较高,表明了较为优良的生物学性能(生物相容性或生物活性)。
上述虽然对本发明的具体实施方式进行了描述,但并非对本发明保护范围的限制,所属领域技术人员应该明白,在本发明的技术方案的基础上,本领域技术人员不需要付出创造性劳动即可做出的各种修改或变形仍在本发明的保护范围以内。
- 还没有人留言评论。精彩留言会获得点赞!