一种基于COF-金属界面协同作用电催化还原二氧化碳的方法与流程
本发明属于多孔晶态材料能源催化应用领域,具体涉及一种具有高化学稳定性的基于c-n单键连接的晶态多孔材料与金属电极复合构成协同界面共同催化二氧化碳电还原的应用。
背景技术
电催化二氧化碳还原是一种高效、可持续的能源转化方式,其可将易获得的电能转化为化学能以便于转化、储存与利用[chem.rev.2010,110,6474;chem.soc.rev.2014,43,631-675;adv.mater.2016,28,3423-3452]。
然而对于金属催化体系,由于金属中较为平缓的能带结构,使得其虽具有较高的催化活性,但并没有专一的选择性,尤其在水体系的二氧化碳电催化还原过程中,氢析出反应相对于二氧化碳还原反应要更胜一筹;另一方面,对于分子催化体系,由于其前线轨道与特定分子(如二氧化碳)匹配较好,使得其具有较好的反应选择性,但受制于催化活性中心的限制,以及分子催化体系化合物本身的导电性影响,使得其效率不高。
技术实现要素:
针对现有催化体系的不足,本发明提出了通过将一种富含-nh-基团的多孔共价有机框架材料(cof)与金属电极相复合,形成一种协同作用的界面,在未改变金属电极费米能级的前提下实现了催化活性和选择性的大幅提升,为电催化二氧化碳还原相关研究提出了一种全新方法。在单独使用分子催化剂或基于分子催化剂的聚合物、金属有机框架材料(mof)、共价有机框架材料(cof)作为电催化剂的方法中,电子需要通过分子骨架抵达催化中心,再对二氧化碳分子进行催化转化,因此二氧化碳电还原反应的效率受到分子基材料的导电性的限制。不同的是,在这里采用的协同方法中电子无需通过有机框架,而是在金属电极与cof材料形成的界面上即可与二氧化碳分子作用使其还原,二氧化碳电还原反应的效率不会因cof材料的导电性而受到限制;而该方法相对于将有机胺类小分子修饰在电极表面的方法,由于cof材料具有较好的晶态结构与有序的孔道结构,其对于二氧化碳传质扩散到电极表面有较积极的作用,在改善选择性的同时并没有阻止二氧化碳分子扩散到电极表面,从而实现了对传统金属电极的改性高效催化二氧化碳电还原。
本发明的技术方案具体如下:
一种基于cof-金属界面协同作用电催化还原二氧化碳的方法,包括以下步骤:
(1)将活化的cof-300在硼氢化钠-甲醇体系中还原,抽滤,固体依次用甲醇和热水洗涤,然后在索氏提取器中用乙醇活化,再在室温下脱气,得到基于仲胺基键合的共价有机框架材料cof-300-ar;
(2)将cof-300-ar粉末与水、乙醇、nafion溶液一起混合,超声分散,形成cof-300-ar分散液;将cof-300-ar分散液缓慢滴加在金属箔上,置于红外灯下烘干,得到cof-金属电极;
(3)以cof-金属电极为工作电极、碳布电极为对电极、饱和甘汞电极为参比电极,以nafion膜作为分离池中的阳离子交换膜,加入新鲜配制的碳酸氢钾溶液作为电解液,组装好电解池后,打开二氧化碳气路向电解液中通气至饱和,然后进行恒电压电解。
步骤(1)的具体步骤为:将1当量对苯二甲酸、含1当量亚胺键的活化的cof-300粉末加入甲醇中,混合均匀;接着于-25~-5℃搅拌条件下缓慢加入不少于38当量的硼氢化钠粉末,在-25~-5℃下继续搅拌0.5~2小时,然后在室温下搅拌至反应充分;反应完成后将反应体系抽滤,依次用甲醇与热水洗涤固体粗产物,除去副产物及催化剂,得到淡黄色粉末;在索氏提取器中将淡黄色粉末用乙醇活化,然后在室温下脱气,得到基于仲胺基键合的共价有机框架材料cof-300-ar。
步骤(2)中,水、乙醇的体积比为3:1。
步骤(2)中,cof-300-ar分散液中,nafion的浓度为0.2%,cof-300-ar的浓度为5g/l。
所述的金属箔为银箔。
步骤(2)中,cof-金属电极上cof-300-ar层的厚度为10-20微米,优选15微米。
步骤(3)中,碳酸氢钾溶液的浓度为0.1m。
本发明具有以下优点和有益效果:
(1)本发明提供cof-金属电极界面协同电催化还原二氧化碳的方法,相较于金属催化以及分子催化体系,既保证了催化活性,也提高了选择性。
(2)构建了多孔材料-金属电极结合的界面,为催化研究拓宽了新思路。
(3)本发明制备工艺简便,易于调节,是一种设计新型电催化电极的有效方法。
附图说明
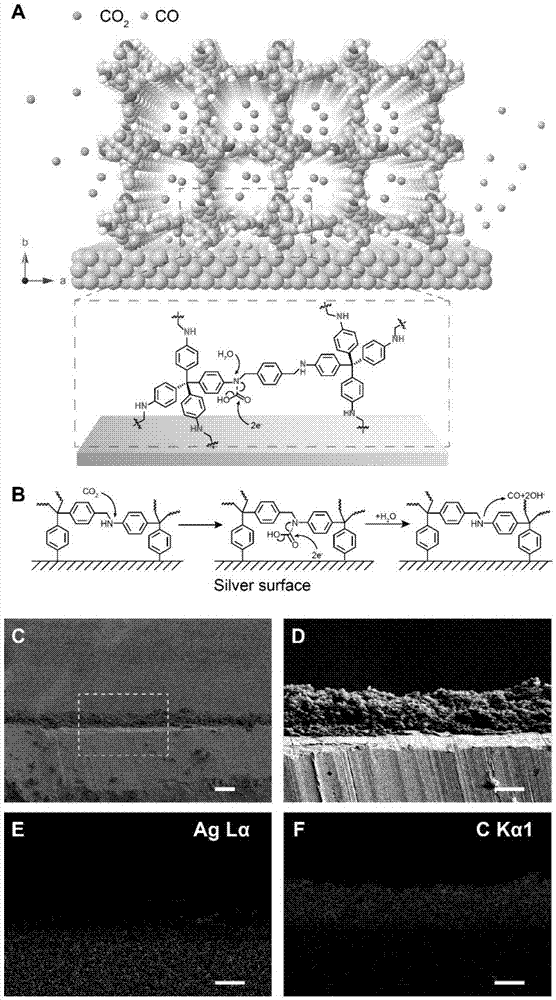
图1为cof-金属电极界面的构建图;其中,图1(a)为cof-金属电极界面的构建示意图,图1(b)展示了cof-金属电极界面对二氧化碳的催化机理,图1(c-f)为扫描电子显微镜与edx能谱对cof-金属电极界面的表征图。
图2为性能测试与机理研究图;其中,图2(a)代表cof-300-ar在两种温度下的二氧化碳吸附等温线图,图2(b)代表cof-300-ar与同位素标记的二氧化碳分子作用后固体核磁共振碳谱表征,图2(c)和图2(d)为cof-银电极与普通银箔、仅涂nafion膜的银箔的性能对比图,分别展示了氢气、二氧化碳的法拉第效率。
图3为cof-300-ar与银电极形成的协同界面的催化机理示意图。
具体实施方式
实施例1
1.基于亚胺键合的三维共价有机框架材料(cof-300)的合成与活化
沿用文献[j.am.chem.soc.,2009,131,4570-4571]中的方法,将100mg四氨基苯基甲烷与64mg对苯二甲醛溶于2ml1,4-二氧六环中,然后加入到玻璃管内,并将玻璃管中的气氛交换为氩气,在液氮冷冻后,进行封管处理;将玻璃管置于120℃烘箱中反应3天,然后滤取固体,将所得固体在索氏提取器中用1,4-二氧六环和丙酮的混合溶剂进行回流活化,然后用超临界态二氧化碳进行活化,得到黄色基于亚胺键合的共价有机框架材料,即活化的cof-300粉末。
2.基于胺键合的cof-300-ar的制备
将57.6mg(1当量)对苯二甲酸、50mg(1当量,以亚胺键数量计)活化的cof-300粉末加入25ml甲醇中,于-15℃下搅拌5分钟;然后在10分钟内分少量多次缓慢加入硼氢化钠粉末共计0.5g(38当量),并继续在-15℃下搅拌1小时,然后在室温下搅拌12小时使反应充分进行;反应完成后将反应液抽滤,依次用甲醇与热水洗涤,除去副产物及催化剂,得到淡黄色粉末;在索氏提取器中将淡黄色粉末用乙醇活化,然后在室温下脱气,得到活化的cof-300-ar粉末样品。
实施例2:cof-金属电极的制备
将5mgcof-300-ar粉末置于0.75ml去离子水、0.25ml乙醇以及40μl5%nafion溶液形成的混合溶液体系中,超声分散5分钟,然后取100μl分散液,缓慢滴加于银箔(购买自alfaaesar)上,置于红外灯下烘干,得到cof-银电极。
实施例3
对cof-300-ar和银箔构建的cof-银电极上的cof-金属界面进行扫描电镜、edx能谱表征,如图1(c-f)所示,发现cof-金属界面清晰、形貌较为均匀,cof-300-ar层的厚度约为15微米,其界面的构建示意图如图1(a)所示。
同时应用谱学技术对cof-300-ar与二氧化碳分子的作用方式进行表征,发现了其中含有类似酰胺的结构,使我们确信cof-300-ar在协同界面中具有富集、传递二氧化碳的作用,进一步体现了这种协同界面在催化反应中的优势。
将cof-300-ar与同位素标记的二氧化碳进行作用,并通过傅里叶变换红外光谱以及固态核磁共振碳谱表征对cof-金属电极催化体系的机理推测与研究,机理推测如图1(b)所示。
实施例4
以cof-银电极为工作电极、碳布电极为对电极、饱和甘汞电极为参比电极,以nafion膜作为分离池中的阳离子交换膜,加入新鲜配制的0.1m碳酸氢钾溶液作为电解液,组装好电解池后,打开二氧化碳气路向电解液中通气至饱和(5分钟),然后进行恒电压电解。
使用上海辰华chi-600电化学工作站控制电势,先进行循环伏安扫描(相对于可逆氢电极,0v~-1.0v),再进行恒电势电解(相对于可逆氢电极,-0.7v或-0.85v),电解进行700秒时对气相色谱(岛津gc-2100plus)进样,通过气相色谱测得的气体含量对反应产物进行分析。发现在两种电势下(相对于可逆氢电极,-0.7v与-0.85v),氢析出反应的法拉第效率分别为22%与9%,而二氧化碳还原为一氧化碳的法拉第效率分别高至53%与80%。
实施例5
本实施例提供一种电催化还原二氧化碳的方法,与实施例4的不同仅在于采用普通银箔代替cof-银电极。
实施例6
本实施例提供一种电催化还原二氧化碳的方法,与实施例4的不同仅在于采用滴加了nafion溶液的普通银箔替代cof-银电极。
结合实施例4-6,通过法拉第效率判断实施例4-6的催化分解效果,结果如图2(c)和图2(d)所示,实施例4的催化分解效果远远好于实施例5和6。
通过以上实施例可知,cof-银电极在电催化二氧化碳还原的反应中,在-0.7v与-0.85v(相对于可逆氢电极)的电势范围下,使得相较于普通银箔,氢气的法拉第效率明显降低,而一氧化碳的法拉第效率显著增加,即催化反应的活性和选择性都获得了较大的提升。
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!