一种E型β-三氟甲基烯酰胺化合物及其电化学氧化合成方法
一种e型
β-三氟甲基烯酰胺化合物及其电化学氧化合成方法
技术领域
1.本说明书一个或多个实施例涉及电化学有机合成技术领域,尤其涉及一种e型β-三氟甲基烯酰胺化合物及其电化学氧化合成方法。
背景技术:
2.作为一类重要的含氟官能团,三氟甲基官能团具备许多独特的性质,例如强吸电子性、良好的代谢稳定性和较好的脂溶性。众多的医药、农用化学品、染料、以及许多功能材料中都含有三氟甲基官能团。因此,向化合中引入三氟甲基官能团,一直受到化学家们的广泛关注。另一方面,烯酰胺类化合物作为有机合成中一类重要的合成砌块,被广泛用于许多手性胺和含氮杂环化合物的合成。对烯酰胺的β-c(sp2)-h直接官能团化反应是构建官能团化烯酰胺化合物的一种有效策略,至此,已取得系列重要研究成果。然而,有关合成β-三氟甲基烯酰胺化合物的报道却相对较少。2012年,loh课题组报道了首例铜催化下烯酰胺的β-c(sp2)-h的三氟甲基化反应(c. feng and t.-p.loh,chem.sci.,2012,3,3458.),该反应具有较好的立体选择性,但反应需要使用昂贵的togni’s试剂作为三氟甲基源,且该方法所使用的铜催化剂有毒且制备复杂。2021年,yang课题组报道了一例光催化下烯酰胺的β-c(sp2)-h的三氟甲基化反应(k.tang,y.chen,j.guan,z. wang,k.chen,h.xiang and h.yang,org.biomol.chem.,2021,19,7475.),该反应在一定程度上避免了有毒化学试剂的使用,但是反应需要价格昂贵的铱催化剂作为光敏剂,且反应立体选择性较差,综上所述,本技术现提出一种e型β-三氟甲基烯酰胺化合物及其电化学氧化合成方法来解决上述出现的问题。
技术实现要素:
3.有鉴于此,本说明书一个或多个实施例的目的在于提出一种e型β-三氟甲基烯酰胺化合物及其电化学氧化合成方法,以解决背景技术中提出的问题。
4.基于上述目的,本说明书一个或多个实施例提供了一种e型β-三氟甲基烯酰胺化合物,具有式(i)所示结构:
[0005][0006]
其中,r1为氢、烷基、烷氧基、卤素、羟基、芳基、酰基或酯基等;r2为苄基、取代苄基、甲基、酰基或叔丁氧羰基。
[0007]
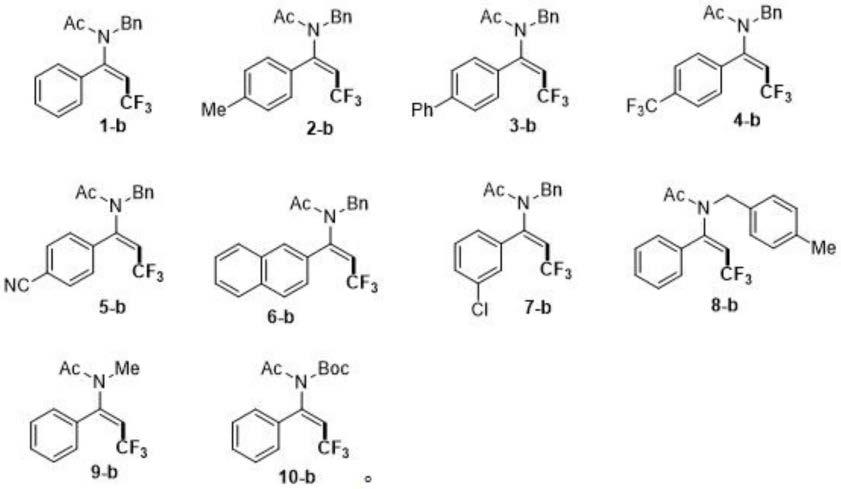
优选的,所述e型β-三氟甲基烯酰胺化合物包括:
[0008][0009]
一种电化学氧化合成方法,用于合成前文所述的e型β-三氟甲基烯酰胺化合物,包括以下步骤:
[0010]
在装配有电极片的反应器中,将非环烯酰胺类化合物、三氟甲基亚磺酸盐、添加剂和溶剂进行搅拌混合,在电化学条件下,进行β-c(sp2)-h三氟甲基化反应,高选择性地得到了系列e型β-三氟甲基烯酰胺化合物。
[0011]
更为优选的,所述非环烯酰胺类化合物具有式(ii)所示的结构:
[0012][0013]
更为优选的,所述三氟甲基亚磺酸盐具有式(ⅲ)所式结构:
[0014]
cf3so2m
[0015]
式(iii);
[0016]
其中,m为金属阳离子。
[0017]
更为优选的,所述添加剂选自甲酸、乙酸、硫酸、盐酸、三氟乙酸和磷酸中的任意一种或几种混合。
[0018]
更为优选的,所述溶剂选自二甲基亚砜、n,n-二甲基甲酰胺、n,n
‑ꢀ
二甲基乙酰胺、乙腈、甲醇、乙醇、四氢呋喃和二氯甲烷中的任意一种或几种混合。
[0019]
更为优选的,所述非环烯酰胺类化合物、三氟甲基亚磺酸盐、添加剂的摩尔比为1.0:(1.0~5.0):(0.5~3.0);所述非环烯酰胺类化合物的起始浓度为0.02~0.1mol/l。
[0020]
更为优选的,所述电化学条件中使用的反应器为不分开电解槽,电源为直流稳压电源,反应在恒定电流的条件下进行,通电电流为5~15ma。
[0021]
更为优选的,所述三氟甲基化反应温度为0~60℃;反应时间为2~10h。
[0022]
更为优选的,所述三氟甲基化反应还包括分离提纯;所述分离提纯的方式选自柱层析色谱、液相色谱、蒸馏和重结晶中的任意一种或多种。
[0023]
从上面所述可以看出,本发明包括以下有益效果:
[0024]
从上面所述可以看出,本发明的有益效果:提供了一种电化学氧化合成 e型β-三氟甲基烯酰胺化合物的方法。在电化学条件下,进行β-c(sp2)
ꢀ‑
h三氟甲基化反应,高选择性地得到了系列e型β-三氟甲基烯酰胺化合物。本发明采用电流为氧化剂,无需额外添加电解质,也无需加入各类金属催化剂和化学氧化剂;反应条件温和,底物普适性广,官能团耐受性好;本发明简单易行,反应体系简单,污染小,符合绿色化学理念,具有良好的应用潜力。
附图说明
[0025]
为了更清楚地说明本说明书一个或多个实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本说明书一个或多个实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0026]
图1-图3为本发明中实施例1制备的产物的核磁氢谱、碳谱和氟谱图;
[0027]
图4-图6为本发明中实施例2制备的产物的核磁氢谱、碳谱和氟谱图;
[0028]
图7-图9为本发明中实施例3制备的产物的核磁氢谱、碳谱和氟谱图;
[0029]
图10-图12为本发明中实施例4制备的产物的核磁氢谱、碳谱和氟谱图;
[0030]
图13-图15为本发明中实施例5制备的产物的核磁氢谱、碳谱和氟谱图;
[0031]
图16-图18为本发明中实施例6制备的产物的核磁氢谱、碳谱和氟谱图;
[0032]
图19-图21为本发明中实施例7制备的产物的核磁氢谱、碳谱和氟谱图;
[0033]
图22-图24为本发明中实施例8制备的产物的核磁氢谱、碳谱和氟谱图;
[0034]
图25-图27为本发明中实施例9制备的产物的核磁氢谱、碳谱和氟谱图;
[0035]
图28-图30为本发明中实施例10制备的产物的核磁氢谱、碳谱和氟谱图。
具体实施方式
[0036]
为使本公开的目的、技术方案和优点更加清楚明白,以下结合具体实施例,对本公开进一步详细说明。
[0037]
本说明书一个或多个实施例提供了一种e型β-三氟甲基烯酰胺化合物,具有式(i)所示结构:
[0038][0039]
其中,r1为氢、烷基、烷氧基、卤素、羟基、芳基、酰基或酯基等;r2为苄基、取代苄基、甲基、酰基或叔丁氧羰基。
[0040]
作为上述方案的改进方案,所述e型β-三氟甲基烯酰胺化合物包括:
[0041][0042]
本发明实施例中提出的一种电化学氧化合成方法,用于合成前文所述的 e型β-三氟甲基烯酰胺化合物,包括以下步骤:
[0043]
在装配有电极片的反应器中,将非环烯酰胺类化合物、三氟甲基亚磺酸盐、添加剂和溶剂进行搅拌混合,在电化学条件下,进行β-c(sp2)-h三氟甲基化反应,高选择性地得到了系列e型β-三氟甲基烯酰胺化合物。
[0044]
作为上述方案的改进方案,所述非环烯酰胺类化合物具有式(ii)所示的结构:
[0045][0046]
作为上述方案的改进方案,所述三氟甲基亚磺酸盐具有式(ⅲ)所式结构:
[0047]
cf3so2m
[0048]
式(iii);
[0049]
其中,m为金属阳离子。
[0050]
作为上述方案的改进方案,所述添加剂选自甲酸、乙酸、硫酸、盐酸、三氟乙酸和磷酸中的任意一种或几种混合。
[0051]
作为上述方案的改进方案,所述溶剂选自二甲基亚砜、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、乙腈、甲醇、乙醇、四氢呋喃和二氯甲烷中的任意一种或几种混合。
[0052]
作为上述方案的改进方案,所述非环烯酰胺类化合物、三氟甲基亚磺酸盐、添加剂的摩尔比为1.0:(1.0~5.0):(0.5~3.0);所述非环烯酰胺类化合物的起始浓度为0.02~0.1mol/l,进一步优选为0.04~0.08mol/l。
[0053]
作为上述方案的改进方案,所述电化学条件中使用的反应器为不分开电解槽,电源为直流稳压电源,反应在恒定电流的条件下进行,通电电流为5~15 ma,进一步优选为8~12ma。
[0054]
作为上述方案的改进方案,所述三氟甲基化反应温度为0~60℃;进一步优选为20~40℃;反应时间为2~10h,进一步优选为4~8h。
[0055]
在本发明中,所述e型β-三氟甲基烯酰胺化合物的制备过程如下所示:
[0056][0057]
本发明中对于所述电极没有特殊限定,可以为铂电极、碳电极、镍电极、铁电极、铜电极、镁电极等本领域技术人员熟知的电极。
[0058]
本发明对于搅拌混合的方式没有特殊限定,采用本领域技术人员熟知的搅拌混合的方式即可。
[0059]
所述三氟甲基化反应还包括分离提纯;所述分离提纯的方式优选为柱层析色谱;所述柱层析色谱的洗脱剂优选但并不限于石油醚与乙酸乙酯的混合溶剂。
[0060]
作为上述方案的改进方案,所述三氟甲基化反应还包括分离提纯;所述分离提纯的方式选自柱层析色谱、液相色谱、蒸馏和重结晶中的任意一种或多种。
[0061]
实施例1
[0062]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式1-a所示) (0.3mmol,75.4mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n, n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×ꢀ
15ml)萃取。用15ml饱和食盐水洗涤合并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为12:1)得到77.8mg目标产物(式1-b所示),收率为81%。
[0063]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图1、图2和图3所示,结构表征数据如下所示:
[0064]1hnmr(400mhz,cdcl3)δ7.49-7.41(m,,3h),7.34-7.28(m,5h),7.17-7.15(m,2h), 5.46(q,j=8.1hz,1h),4.52(s,2h),2.25(s,3h);
13
cnmr(101mhz,cdcl3)δ169.8,149.7 (q,j=5.9hz),136.4,132.8,130.6,128.8(q,j=2.1hz),128.49,128.51,128.4,127.5,122.0 (q,j=270.2hz),116.7(q,j=34.9hz),49.4,22.4;
19
f nmr(377mhz,cdcl3)δ-55.94.
[0065][0066]
实施例2
[0067]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式2-a所示) (0.3mmol,79.6mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n, n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×ꢀ
15ml)萃取。用15ml饱和食盐水洗涤合
并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为12:1)得到69.1mg目标产物(式2-b所示),收率为69%。
[0068]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图4、图5和图6所示,结构表征数据如下所示:
[0069]1hnmr(400mhz,cdcl3)δ7.32-7.26(m,3h),7.24(s,4h),7.19-7.16(m,2h),5.41 (q,j=8.2hz,1h),4.53(s,2h),2.41(s,3h),2.25(s,3h);
13
c nmr(101mhz,cdcl3)δ 170.0,149.8(q,j=5.9hz),141.1,136.6,129.9,129.4,128.8(q,j=2.0hz),128.6,128.5, 127.6,122.2(q,j=270.2hz),116.3(q,j=35.0hz),49.5,22.5,21.4;
19
f nmr(377mhz, cdcl3)-55.92.
[0070][0071]
实施例3
[0072]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式3-a所示) (0.3mmol,98.2mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n,n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×ꢀ
15ml)萃取。用15ml饱和食盐水洗涤合并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为12:1)得到87.7mg目标产物(式3-b所示),收率为74%。
[0073]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图7、图8和图9所示,结构表征数据如下所示:
[0074]1h nmr(400mhz,cdcl3)δ7.69-7.63(m,4h),7.51-7.46(m,2h),7.45-7.40(m, 3h),7.36-7.27(m,3h),7.24-7.20(m,2h),5.50(q,j=8.2hz,1h),4.60(s,2h),2.30(s, 3h);
13
c nmr(101mhz,cdcl3)δ170.0,149.5(q,j=5.7hz),143.5,139.7,136.5,131.5, 129.3(q,j=2.0hz),128.9,128.6,128.5,128.0,127.6,127.2,127.1,122.1(q,j=270.1hz), 116.7(q,j=35.2hz),49.6,22.5;
19
f nmr(377mhz,cdcl3)-55.88.
[0075][0076]
实施例4
[0077]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式4-a所示) (0.3mmol,95.8mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n, n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×ꢀ
15ml)萃取。用15ml饱和食盐水洗涤合
并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为12:1)得到72.1mg目标产物(式4-b所示),收率为62%。
[0078]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图10、图11和图12所示,结构表征数据如下所示:
[0079]1h nmr(400mhz,cdcl3)δ7.67(d,j=8.1hz,2h),7.42(d,j=8.1hz,2h),7.34
‑ꢀ
7.23(m,3h),7.13(dd,j=7.6,1.9hz,2h),5.58(q,j=8.0hz,1h),4.53(s,2h),2.26(s,3h);
13
c nmr(101mhz,cdcl3)δ169.8,148.4(q,j=6.0hz),136.6,136.1,132.7,132.3,129.3 (q,j=1.8hz),128.7,128.4,127.9,125.6(q,j=3.8hz),122.7(q,j=99.3hz),118.1(q,j= 35.0hz,1h),49.8,22.5;
19
f nmr(377mhz,cdcl3)-55.93,-63.00.
[0080][0081]
实施例5
[0082]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式5-a所示) (0.3mmol,82.9mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n, n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×ꢀ
15ml)萃取。用15ml饱和食盐水洗涤合并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为10:1)得到68.2mg目标产物(式5-b所示),收率为66%。
[0083]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图13、图14和图15所示,结构表征数据如下所示:
[0084]1h nmr(400mhz,cdcl3)δ7.72-7.68(m,2h),7.40(d,j=8.3hz,2h),7.33-7.27 (m,3h),7.10(dd,j=7.4,2.2hz,2h),5.61(q,j=8.0hz,1h),4.54(s,2h),2.25(s,3h);
13
c nmr(101mhz,cdcl3)δ169.8,148.0(q,j=5.9hz),137.5,135.9,132.2,129.4(q,j=2.0 hz),128.7,128.2,127.9,121.7(q,j=270.6hz),118.3(q,j=35.3hz),117.8,114.3,50.0, 22.5;
19
f nmr(377mhz,cdcl3)-55.86.
[0085][0086]
实施例6
[0087]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式6-a所示) (0.3mmol,90.4mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n, n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×ꢀ
15ml)萃取。用15ml饱和食盐水洗涤合
并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为14:1)得到60.9mg目标产物(式6-b所示),收率为55%。
[0088]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图16、图17和图18所示,结构表征数据如下所示:
[0089]1hnmr(400mhz,cdcl3)δ7.92-7.82(m,4h),7.62-7.52(m,2h),7.41(dd,j=8.5, 1.8hz,1h),7.35-7.26(m,3h),7.19(dd,j=7.8,1.8hz,2h),5.57(q,j=8.2hz,1h),4.58 (s,2h),2.32(s,3h);
13
c nmr(101mhz,cdcl3)δ170.1,149.8(q,j=5.9hz),136.5,134.1, 132.6,130.1,129.4,128.6,128.5,127.7,127.7,127.7,126.9,125.2,122.2(q,j=270.3hz), 116.8(q,j=35.0hz),49.7,22.6;
19
f nmr(377mhz,cdcl3)-55.76.
[0090][0091]
实施例7
[0092]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式7-a所示) (0.3mmol,85.7mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n, n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×ꢀ
15ml)萃取。用15ml饱和食盐水洗涤合并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为12:1)得到99.8mg目标产物(式7-b所示),收率为94%。
[0093]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图19、图20和图21所示,结构表征数据如下所示:
[0094]1hnmr(400mhz,cdcl3)δ7.45(ddd,j=8.0,2.0,1.1hz,1h),7.36(t,j=7.9hz,1h), 7.33-7.25(m,4h),7.23-7.19(m,1h),7.14(dd,j=7.7,1.8hz,2h),5.50(q,j=8.0hz, 1h),4.52(s,2h),2.25(s,3h);
13
c nmr(101mhz,cdcl3)δ169.8,148.3(q,j=5.9hz), 136.2,134.7,130.7,129.8,128.6128.4,127.7,127.2,127.2,121.8(q,j=270.4hz),117.6(q, j=35.0hz),49.6,22.4;
19
f nmr(377mhz,cdcl3)-55.95.
[0095][0096]
实施例8
[0097]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式8-a所示) (0.3mmol,79.6mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n, n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×ꢀ
15ml)萃取。用15ml饱和食盐水洗涤合
并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为12:1)得到73.1mg目标产物(式8-b所示),收率为73%。
[0098]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图22、图23和图24所示,结构表征数据如下所示:
[0099]1h nmr(400mhz,cdcl3)δ7.50-7.39(m,3h),7.36-7.31(m,2h),7.10(d,j=7.9 hz,2h),7.05(d,j=8.0hz,2h),5.45(q,j=8.2hz,1h),4.48(s,2h),2.32(s,3h),2.24(s, 3h);
13
c nmr(101mhz,cdcl3)δ169.9,149.8(q,j=5.9hz),137.3,133.4,132.9,130.7, 129.2,128.9,128.6,128.6,122.1(q,j=270.1hz),116.8(q,j=35.0hz),49.2,22.5,21.1;
19
f nmr(377mhz,cdcl3)-55.86.
[0100][0101]
实施例9
[0102]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式9-a所示) (0.3mmol,52.6mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n, n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×
15ml)萃取。用15ml饱和食盐水洗涤合并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为15:1)得到40.1mg目标产物(式9-b所示),收率为56%。
[0103]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图25、图26和图27所示,结构表征数据如下所示:1h nmr(400mhz,cdcl3)δ7.47-7.37(m,3h),7.37-7.33(m,2h),5.69(q,j=8.1hz, 1h),2.94(s,3h),2.14(s,3h);
13
c nmr(101mhz,cdcl3)δ170.3,151.6(q,j=5.7hz), 133.1,130.6,128.6122.4(q,j=269.9hz),114.6(q,j=35.2hz),35.2,22.3;
19
f nmr(377 mhz,cdcl3)-55.73.
[0104][0105]
实施例10
[0106]
向10ml的不分开电解槽中依次加入非环烯酰胺类化合物(式10-a所示) (0.3mmol,74.5mg),三氟甲基亚磺酸钠(0.9mmol,140.4mg),n, n-二甲基甲酰胺(5ml),硫酸(0.45mmol,24.5μl);插入石墨块作为阳极(大小:10mm
×
10mm
×
0.3mm)和铂片作为阴极(大小:10mm
ꢀ×
10mm
×
0.2mm),直流电源供电10ma,在室温下搅拌反应。反应完成后(tlc跟踪监测),将所得反应液倒入15ml水中,用乙酸乙酯(3
×ꢀ
15ml)萃取。用15ml饱和食盐水洗涤合并的有机相。有机相用无水硫酸钠干燥,用旋转蒸发仪浓缩除去溶剂,通过柱层析分离(石油醚:乙酸乙酯的体积比为50:1)得到46.4mg目标产物(式10-b所示),收率为47%。
[0107]
对所得产物的结构进行表征,核磁共振氢谱、核磁共振碳谱和核磁共振氟谱图分别如图28、图29和图30所示,结构表征数据如下所示:1h nmr(400mhz,cdcl3)δ7.43-7.38(m,2h),7.38-7.31(m,3h),5.77(q,j=8.0hz, 1h),2.53(s,3h),1.37(s,9h);
13
c nmr(101mhz,cdcl3)δ172.3,151.4,146.6(q,j=6.6 hz),134.5,129.7,128.7(q,j=2.3hz),127.9,122.1(q,j=270.2hz),119.2(q,j=35.2hz), 84.3,27.6,26.1;
19
f nmr(377mhz,cdcl3)-56.85.
[0108][0109]
本说明书一个或多个实施例旨在涵盖落入所附权利要求的宽泛范围之内的所有这样的替换、修改和变型。因此,凡在本说明书一个或多个实施例的精神和原则之内,所做的任何省略、修改、等同替换、改进等,均应包含在本公开的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1