一种电催化合成1-碘代炔类化合物的方法
1.本发明属于有机合成技术领域,具体涉及一种电催化合成1-碘代炔类化合物的方法。
背景技术:
2.1-碘代炔烃是有机合成的重要中间体,广泛应用于各种高级结构如共轭二炔,烯炔,取代烯烃,杂环和功能聚合物的前体,而且由于它们的独特结构涉及可控的亲电性和亲核性,也被认为是双官能化的分子。现有技术报道了以末端炔烃为原料,采用多种金属催化剂、高价碘盐法、离子液体法、碱法、相转移催化剂法、超声波法、碘氧化物法、格氏试剂法和/或正丁基锂法等方法可以制备1-碘代炔烃。
3.有机电合成是一种高效而温和的合成工具,它可以通过阳极氧化和阴极还原从而在无外源性氧化剂和还原剂的条件下实现氧化还原。环保是电化学合成的优点之一,传统方法通常在高温或高压下进行,而电化学反应通常在较温和的条件下进行。传统反应通常需要淬灭,而电化学反应通过关闭电源开关可以随时停止。由于电化学反应的反应效率高,因此反应时间通常较短,易于扩大规模,在工业应用中具有巨大潜力。电催化技术具有广泛优势和多种用途,而且能够通过催化剂、电解液、界面、电势等进行调控。
4.通过大量的文献调研,现有技术中仅一篇文献公开了电催化合成1-碘代炔的方法(synlett 2000, no. 1, 89
–
91),该方法以甲醇为溶剂、碘化钠为电解质,在7.5ma/cm2电流密度、25℃及氮气环境下制备获得系列1-碘代炔类化合物。然而,该方法一方面需要严格的惰性气氛保护,在空气气氛下进行时目标产物的产率显著降低甚至无法进行;另一方面则溶剂体系为有机溶剂甲醇,原子经济性仍然有待提高。
技术实现要素:
5.本发明的目的在于克服现有技术缺陷,提供一种电催化合成1-碘代炔类化合物的方法。在空气气氛下,炔烃类底物在乙腈和水的混合溶剂体系中以碘化钠为电解质顺利地制备获得系列1-碘代炔类化合物。较之现有技术方法,本发明的技术优势在于无需保护气氛降低了工艺成本、设备要求和操作难度,减少了有机溶剂的用量提高了原子经济性,不使用金属催化剂、碱、氧化剂和/或其它反应助剂,反应条件温和简单易于操作。
6.根据本发明提供的一种电催化合成1-碘代炔类化合物的方法,包括如下步骤:式i所示的炔烃类化合物、碘盐、乙腈和水依次加入反应器中,在空气气氛和室温下,控制电流6-12 ma反应,反应完全后经后处理得到式ii所示的1-碘代炔类化合物。
7.反应式如下:
其中,m表示1,2,3,4,5的整数;各个r取代基相同或不同,彼此独立地选自氢、卤素、-cn、-no2、-oh、-sh、c
1-10
烷基、c
1-10
烷氧基、c
1-10
烷硫基、c
1-10
卤代烷基、c
6-20
芳基、c
1-10
烷基羰基、c
1-10
烷氧羰基;和/或被卤素、-cn、-no2、-oh、-sh、c
1-10
烷基、c
1-10
烷氧基、c
1-10
烷硫基、c
1-10
卤代烷基、c
6-20
芳基、c
1-10
烷基羰基、c
1-10
烷氧羰基取代的c
6-20
芳基;和/或相邻的两个r取代基彼此相连,并与连接这两个r取代基的芳环碳原子共同构成含或不含杂原子的五至七元环状结构单元。
8.优选地,m表示1,2,3,4,5的整数;各个r取代基相同或不同,彼此独立地选自氢、氟、氯、溴、-cn、-no2、-oh、-sh、c
1-3
烷基、c
1-3
烷氧基、c
1-3
烷硫基、c
1-3
卤代烷基、c
6-12
芳基、c
1-3
烷基羰基、c
1-3
烷氧羰基;和/或被卤素、-cn、-no2、c
1-3
烷基、c
1-3
烷氧基、取代的c
6-20
芳基;和/或相邻的两个r取代基彼此相连,并与连接这两个r取代基的芳环碳原子共同构成不含杂原子的五至七元环状结构单元。
9.进一步优选地,m表示1,2,3,4,5的整数;各个r取代基相同或不同,彼此独立地选自氢、氟、氯、溴、-cn、-no2、甲氧基、乙氧基、丙氧基、甲基、乙基、丙基、苯基、乙酰基、叔丁氧羰基;和/或两个r取代基彼此相连,并与连接这两个r取代基的芳环碳原子共同构成苯环结构单元。
10.根据本发明前述的方法,其中所述碘盐为碘化钠或碘化钾,优选为碘化钠。
11.根据本发明前述的方法,其中乙腈和水的体积比为(2.5~8): 1,优选为5:1。
12.根据本发明前述的方法,其中电流优选为8 ma,反应完全需要的时间为2~5小时,优选为3~4小时。
13.根据本发明前述的方法,其中式i所示的炔烃类化合物和碘盐的投料摩尔比为1:(2~4),优选为1:3。
14.根据本发明前述的方法,其中所述后处理操作如下:将反应液转入分液漏斗中,加入乙酸乙酯和饱和硫代硫酸钠溶液,洗涤和萃取分液,有机相经干燥、过滤、浓缩后得到粗产物,将粗产物经柱色谱层析分离得到式ii所示的1-碘代炔类化合物。
15.较之现有技术,本发明的电催化合成1-碘代炔的方法具有如下显著的优势:1)本发明电催化合成1-碘代炔的方法仅使用碘化钠作为电解质,不使用金属催化剂、碱、氧化剂和/或其它反应助剂,反应条件温和简单易于操作,目标产物产率高达88%以上。
16.2)本发明的电催化合成1-碘代炔的方法较之现有技术(synlett 2000, no. 1, 89
–
91)的电催化合成方法而言,本发明不再需要严格的惰性气氛保护,对反应装置、工艺成本及操作的要求显著降低,减少了有机溶剂用量,提高了工艺的原子经济性。
附图说明
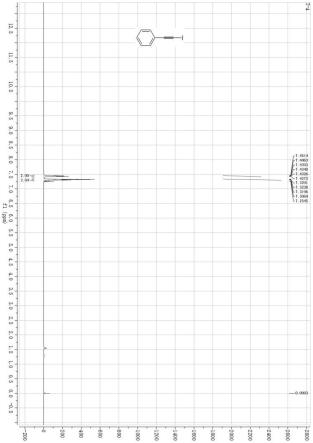
17.图1 为实施例8制备的产物的1h nmr谱图。
18.图2 为实施例8制备的产物的
13
c nmr谱图。
19.图3 为实施例22制备的产物的1h nmr谱图。
20.图4 为实施例22制备的产物的
13
c nmr谱图。
21.图5 为实施例23制备的产物的1h nmr谱图。
22.图6 为实施例23制备的产物的
13
c nmr谱图。
23.图7 为实施例24制备的产物的1h nmr谱图。
24.图8 为实施例24制备的产物的
13
c nmr谱图。
25.图9 为实施例25制备的产物的1h nmr谱图。
26.图10 为实施例25制备的产物的
13
c nmr谱图。
具体实施方式
27.以下结合具体实施例,对本发明作进一步地描述。在下文中,如无特殊说明,所使用的方法均为本领域常规方法,所使用的原料及试剂均是通过常规商业途径购买获得和/或基于有机合成领域经典方法制备获得。
28.实施例1-17 反应条件优化试验以苯乙炔和碘化钠为模板底物,探索了不同电催化合成条件下对于产率的影响,反应式如下:。
29.表1:基本反应条件:苯乙炔0.2mmol,室温,空气。
30.以实施例8为例,典型试验操作如下:在10毫升三口烧瓶中加入碘化钠(0.6 mmol)、苯乙炔(0.2 mmol)、乙腈2.5毫升、水0.5毫升, 室温,空气,电流8 ma,3小时后反应完全, 转入分液漏斗中,加入20毫升乙酸乙酯,用饱和硫代硫酸钠溶液洗涤(10 ml
×
2),将乙酸乙酯有机相用无水mgso4干燥, 过滤后, 减压蒸去溶剂得粗产物, 经柱色谱分离(石油醚洗脱)得到目标产物1-碘-2-苯基乙
炔,产率88%。1h nmr (400 mhz, cdcl3) δ 7.45
ꢀ–ꢀ
7.43 (m, 2h), 7.33
ꢀ–ꢀ
7.31 (m, 3h);
13
c nmr (100 mhz, cdcl3) δ 132.31, 128.79, 128.23, 123.36, 94.12, 6.10。
31.由表1可以看出,溶剂是影响本发明的电催化合成1-碘代炔的方法的主要因素,当溶剂为丙酮/水混合溶剂时,反应无法进行;当溶剂替换为甲醇/水时,则可以获得47%的目标产物产率。有意思的是,当完全采用甲醇作为溶剂时,在空气气氛下进行反应仅获得38%的目标产物产率,这表明现有技术(synlett 2000, no. 1, 89
–
91)的方法需要严格地在惰性气氛条件下才能顺利地实施。经过对溶剂体系、电流大小、反应时间、电解质用量等不同工艺条件的优化,本发明的最优化反应条件为:溶剂选择乙腈:水体积比=5:1、室温、空气气氛、电流8 ma,碘化钠用量为3摩尔当量。
32.实施例18-21 碘盐底物拓展试验在获得了最佳反应条件的基础上,进一步探究了不同碘盐对反应的影响。即仅替换碘盐的种类,在实施例8的操作条件下进行反应,反应式如下:。
33.表2: 碘盐产率18碘化铵0%19碘化钾85%20碘化锌《10%21碘化四丁基铵30%由表2可以看出,碘化铵作为电解质时反应无法进行,碘化钾作为电解质与碘化钠基本相当,而其它的碘金属盐如碘化锌、季铵盐如碘化四丁基铵作为电解质则产率显著降低。
[0034] 实施例22-25 炔烃类底物拓展试验仅替换炔烃类反应底物,在在实施例8的操作条件下进行反应,反应式如下:。
[0035]
表3: 炔烃反应时间温度产率223小时室温91%233小时室温90%243小时室温94%254小时室温88%由表3可以看出,本发明的电催化合成1-碘代炔的方法具有良好的官能团普适性,
适合于制备各种类型的1-碘代炔类化合物。
[0036]
以上所述实施例仅为本发明的优选实施例,而并非本发明可行实施的穷举。对于本领域技术人员而言,在不背离本发明原理和精神的前提下,对其所作出的任何显而易见的改动,都应当被认为包含在本发明的权利要求保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1