生物样本中美他卡韦及其代谢产物浓度的检测方法与流程
本发明涉及生物样本中6-甲氧基-2,3-双脱氧鸟苷及其代谢产物浓度的检测方法,特别涉及一种采用UPLC-MS/MS联用技术分析美他卡韦及其代谢产物2,3-双脱氧鸟苷和O6-甲基鸟嘌呤含量的方法。
背景技术:
目前,乙型肝炎已经成为全球性危害的态势,而长期应用核苷类似药物抗病毒治疗仍是迄今切实可行的治疗方式,6-甲氧基-2,3-双脱氧鸟苷(代号:PNA)属于核苷类抗乙肝病毒感染药物,可能的药理作用机制包括直接作用和间接作用。直接作用是指PNA作为鸟嘌呤类似物能直接进入肝细胞并在细胞内被磷酸化形成有活性的三磷酸盐,该三磷酸盐与HBV多聚酶的天然底物三磷酸脱氧鸟嘌呤核苷竞争,从而抑制HBVDNA复制过程。间接作用是指PNA在血浆内很快代谢为2,3-双脱氧鸟苷(DDG),因此可以作为DDG的前药。DDG在细胞内被磷酸化为活性三磷酸盐ddGTP,通过与多聚酶的结合抑制HBVDNA的复制,导致DNA链合成中止。从目前的药效和毒性研究结果来看,PNA是一个安全有效的抗乙型肝炎病毒的药物。对PNA的临床前研究较多,大鼠灌胃给PNA后在体内迅速达峰且快速消除,似乎不是以原型药物发挥药效作用的,但是查阅国内外文献,人体生物样本中PNA及其代谢产物DDG和O6-甲基鸟嘌呤的含量测定及其药代动力学的研究却未见报道。
技术实现要素:
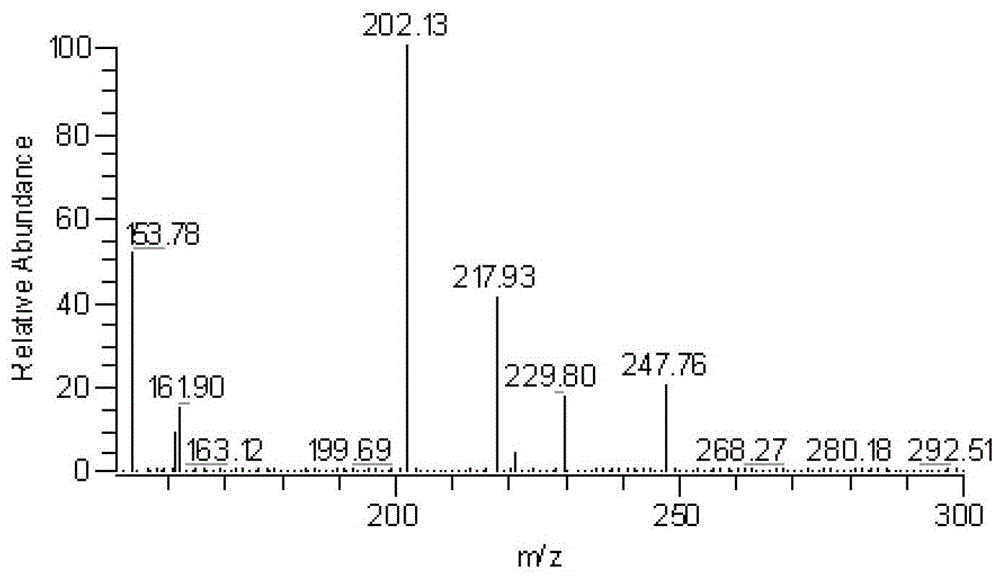
本发明通过对PNA人体中原型及主要代谢产物的研究,建立其血浆、尿样浓度检测方法,为进行PNA人体药代动力学评价、制剂的药学研究、质量标准、工业化生产质控指标的制定和临床方案的制定提供依据。发明人经过大量的实验对比,最终筛选选定了本发明的实验条件。该条件专属性强,可以得到满意的峰形及色谱保留时间,使分析时间相对较短。本发明提供了一种生物样本(血浆、尿)中PNA及其代谢产物DDG和O6-甲基鸟嘌呤浓度的检测方法,包括以下步骤:(a)对生物样品进行前处理:取待测样品,加入内标和乙酸乙酯进行液液萃取,并取上清液吹干,复溶,取上清液,所述的内标为替硝唑溶液,所述的替硝唑的用量为使每1L样品中含替硝唑重量为18-22μg,所述的乙酸乙酯的用量为使每1L样品使用乙酸乙酯体积为3.0-6.0L;(b)采用超高效液相色谱-质谱-质谱(UPLC/MS/MS)联用仪测定上述上清液中PNA、DDG和O6-甲基鸟嘌呤的浓度,其中,UPLC/MS/MS联用的色谱条件是:色谱柱是用十八烷基硅烷键合硅胶为填充剂;内标法,以替硝唑作为内标;流动相为甲醇-醋酸铵水溶液的混合液,其比例范围为(72±4):(28±2),醋酸铵水溶液浓度为每1L水中含醋酸铵重量为7.8-38.5mg,pH为6.5-7.5,UPLC/MS/MS联用的质谱条件是:离子源为ESI离子源,正离子检测。所述步骤(a)中乙酸乙酯体积优选为5L。所述的色谱条件流动相由甲醇-醋酸铵水溶液组成,醋酸铵水溶液最佳浓度为0.2mmol/L,最佳pH为7.0,流动相最佳配比为72:28。所述的UPLC/MS/MS联用质谱条件中,扫描方式为选择性离子检测。所述的生物样品来源于人。附图说明图1A、1B、1C:分别为PNA、DDG和O6-甲基鸟嘌呤的结构式。图2A、2B、2C、2D:分别为PNA、DDG、O6-甲基鸟嘌呤和替硝唑的质谱扫面图。图3A、3B、3C:为实施例一中PNA、DDG、O6-甲基鸟嘌呤和替硝唑的选择性反应检测(SRM)色谱图,Ⅰ、Ⅱ、Ⅲ和Ⅳ分别代表PNA、DDG、替硝唑、O6-甲基鸟嘌呤的色谱峰;其中:图3A代表空白血浆;图3B代表空白血浆中加入PNA(3000ng/mL)、DDG(3000ng/mL)、O6-甲基鸟嘌呤(6000ng/mL)和内标替硝唑(20ng/mL);图3C代表血浆样品。图4A、4B、4C:分别为实施例二中PNA、DDG、O6-甲基鸟嘌呤和替硝唑的选择性反应检测(SRM)色谱图,Ⅰ、Ⅱ、Ⅲ和Ⅳ分别代表PNA、DDG、替硝唑、O6-甲基鸟嘌呤的色谱峰;其中:图4A代表空白尿样;图4B代表空白尿样中加入PNA(3000ng/mL)、DDG(3000ng/mL)、O6-甲基鸟嘌呤(6000ng/mL)和内标替硝唑(18ng/mL);图4C代表尿样样品。具体实施方式为了更加清楚地对本发明进行说明,以下通过具体实施例对本发明的具体实施方式进行更为详细地说明。但是应该理解,以下所述具体实施例仅用于对本发明进行示例性说明,而非用于对本发明进行任何性质的限定,其中所用材料、试剂、仪器及操作条件等仅为代表性的,其并不限于所列举的情况。所属技术领域的技术人员通过阅读以下说明可以对本发明做出不脱离本发明权利要求所限定的保护范围改动和改进,这些改动和改进也处于本发明所要求保护的范围内。实施例一1仪器、材料与试剂1.1仪器Agilent1290超高效液相色谱仪,配有6460型三重四级杆质谱仪,美国Agilent公司。1.2样品与试剂PNA(含量以无水物计为100%),由南京长澳医药科技有限公司提供;DDG购自Sigma;O6-甲基鸟嘌呤(纯度97%),购自Sigma;替硝唑(纯度99.8%),购自中国药品生物制品检定所;乙酸乙酯(HPLC级),购自美国Tedia公司;其它化学试剂均为分析纯。2实验部分2.1血浆样品前处理取10mL玻璃离心管,加1μg/mL内标溶液20μL,加入1mL人血浆,涡旋混匀30s,加5mL乙酸乙酯,涡旋振荡3min,3500rpm离心10min,上清液4mL于37℃水浴中氮气流挥干,用100μL流动相复溶残渣,22000rpm离心10min一次,取上清液10μL进样分析。2.2UPLC-MS/MS分析条件2.2.1色谱条件色谱柱:ThermoBDSHYPERSILC18100*4.6(mm),2.4μm;流动相:0.2mmol/L醋酸铵(pH7.0)溶液:甲醇=72:28;柱温:35℃;流速:0.4mL/min;进样量:10μL。2.2.2质谱条件ESI离子源,正离子选择性反应检测;定量分析的离子分别为:m/z266.0→166.0(PNA)、m/z252.0→152.0(DDG)、m/z166.0→149.0(O6-甲基鸟嘌呤)、m/z248.0→202.0(替硝唑);碰撞能量分别为:5V(PNA)、5V(DDG)、17V(O6-甲基鸟嘌呤)、10V(替硝唑);鞘气流速:35L/min;雾化气流速:15L/min,毛细管温度:350℃;蒸发气温度:300℃;喷雾电压:3500V。2.3UPLC-MS/MS分析结果对样品PNA、DDG、O6-甲基鸟嘌呤和内标替硝唑的质谱扫描图参见图2A、2B、2C。3方法确证3.1专属性空白血浆、空白血浆中加入PNA(3000ng/mL)、DDG(3000ng/mL)、O6-甲基鸟嘌呤(6000ng/mL)和内标替硝唑(20ng/mL)、血浆样品的色谱图分别见图3A、3B和3C,从图中可以看出,空白血浆中的内源性物质不干扰PNA、DDG、O6-甲基鸟嘌呤和内标的测定,内标替硝唑、PNA、DDG、O6-甲基鸟嘌呤的保留时间分别为3.7min、5.1min、2.5min和5.2min。3.2标准曲线PNA、DDG、O6-甲基鸟嘌呤的标准系列溶液由甲醇配制。取空白血浆1mL,加入内标溶液20μL,再依次加入PNA、DDG、O6-甲基鸟嘌呤标准系列溶液,配制成相当于PNA浓度为0.1、0.2、0.5、1、3、10、30、100、300、1000、3000ng/mL,DDG浓度为0.1、0.2、0.5、1、3、10、30、100、300、1000、3000ng/mL,O6-甲基鸟嘌呤浓度为0.2、0.4、1、2、6、20、60、200、600、2000、6000ng/mL的血浆样品,按2.1项下操作,进行UPLC-MS/MS分析,用加权(W=1/x2)最小二乘法进行回归运算,根据标准曲线,本方法PNA的线性范围为0.1~3000ng/mL,标准曲线的方程为:y=0.1621x-0.02033(r=0.9945),DDG的线性范围为0.1~3000ng/mL,标准曲线的方程为:y=0.001648x-0.003261(r=0.9974),O6-甲基鸟嘌呤的线性范围为0.2~6000ng/mL,标准曲线的方程为:y=0.1106x-0.05448(r=0.9947),y表示待测物与内标的峰面积之比,x表示待测物的浓度。3.3提取回收率取人空白血浆1mL,按3.2项下操作,制备低、中、高三个浓度(PNA0.2、10、1000ng/mL;DDG0.2、10、1000ng/mL、O6-甲基鸟嘌呤0.4、20、2000ng/mL)的质控样品,每个浓度进行5样本分析,记录色谱峰。同时,分别取与低、中、高三种浓度水平时等量的标准液,置10mL具塞离心管中,每种水平样品各2份,各精密加入内标溶液20μL(1μg/mL),于37℃水浴中氮气流吹干,残留物以100μLUPLC-MS/MS测定用流动相涡旋溶解,22000rpm离心10min。吸取上清液10μL进样分析,获得相应峰面积,以二者色谱图的峰面积之比,考察样品的提取回收率。测定和分析结果见表1-1~1-3。表1-1PNA的提取回收率(n=5)表1-2DDG的提取回收率(n=5)表1-3O6-甲基鸟嘌呤的提取回收率(n=5)如表1-1~1-3所示,PNA三个浓度的提取回收率分别为65.2%、65.6%、63.8%。DDG的提取回收率分别为57.4%、61.7%、60.5%。O6-甲基鸟嘌呤的提取回收率分别为63.7%、63.1%、64.4%。3.4精密度与准确度按3.2项下操作,制备PNA、DDG、O6-甲基鸟嘌呤低、中、高三个浓度的质量控制样品,每个浓度进行5样本分析,连续测定三天,方法精密度由求得的日内、日间RSD(%)来评价,准确度由实际测得值与理论值比值来评价,分析结果如表2-1~2-3所示。表2-1血浆样品中PNAUPLC-MS/MS测定方法的准确度与精密度(每浓度每天5样品,连续3天)表2-2血浆样品中DDGUPLC-MS/MS测定方法的准确度与精密度(每浓度每天5样品,连续3天)表2-3血浆样品中O6-甲基鸟嘌呤UPLC-MS/MS测定方法的准确度与精密度(每浓度每天5样品,连续3天)如表2-1~2-3所示,PNA日内、日间精密度分别在9.2%及8.3%以内,准确度范围为89.1~102.7%;DDG日内、日间精密度分别在7.9%及8.1%以内,准确度范围为91.4~99.2%;O6-甲基鸟嘌呤日内、日间精密度分别在8.3%及9.0%以内,准确度范围为91.3~105.2%。表明本方法具有良好的精密度与准确度。3.5稳定性以-20℃冷冻10天及冷冻-溶化三次来考察PNA、DDG、O6-甲基鸟嘌呤的稳定性,如果实测值与理论值的偏差在±15%以内,则表明样品稳定,本实验方法的稳定性结果见表3-1~3-3。表3-1PNA稳定性结果(n=3)表3-2DDG稳定性结果(n=3)表3-3O6-甲基鸟嘌呤稳定性结果(n=3)如表3-1~3-3所示,PNA、DDG、O6-甲基鸟嘌呤浓度的实测值与理论值的偏差在±15%以内,表明本方法具有良好的稳定性。3.6基质效应取空白血浆1mL共15份(5种不同来源、每种三份),置10mL玻璃离心管中,加入乙酸乙酯5mL,涡旋振荡3min。3500rpm离心10min,取上清液4mL于另一玻璃离心管,分别加入PNA、DDG、O6-甲基鸟嘌呤标准液适量,配成低、中、高三个浓度水平,每种水平样品各5份,各精密加入内标溶液20μL(1μg/mL),涡旋混匀后37℃水浴中氮气流吹干,用100μL流动相复溶残渣,22000rpm离心10min一次,取上清液10μL进样分析。另分别取与基质样品组低、中、高三个浓度水平时等量的PNA、DDG、O6-甲基鸟嘌呤标准液,置10ml具塞离心管中,每种水平样品各5份,各精密加入内标溶液20μL(1μg/mL),于37℃水浴中氮气流吹干,用100μL流动相复溶残渣,22000rpm离心10min一次,取上清液10μL进样分析。结果表明基质对两者离子化效率的影响基本稳定(表4-1~4-3)。表4-1PNA基质效应结果(n=5)浓度(ng/mL)基质效应(%)RSD(%)0.289.6±8.08.91094.3±6.77.11000102.7±6.15.9表4-2DDG基质效应结果(n=5)浓度(ng/mL)基质效应(%)RSD(%)0.291.4±8.69.41089.7±5.56.11000104.5±5.65.4表4-3O6-甲基鸟嘌呤基质效应结果(n=5)浓度(ng/mL)基质效应(%)RSD(%)0.493.7±7.98.420104.2±7.26.9200095.1±4.54.7实施例二1仪器、材料与试剂1.1仪器Agilent1290超高效液相色谱仪,配有6460型三重四级杆质谱仪,美国Agilent公司。1.2样品与试剂PNA(含量以无水物计为100%),由南京长澳医药科技有限公司提供;DDG购自Sigma;O6-甲基鸟嘌呤(纯度97%),购自Sigma;替硝唑(纯度99.8%),购自中国药品生物制品检定所;乙酸乙酯(HPLC级),购自美国Tedia公司;其它化学试剂均为分析纯。2实验部分2.1尿样前处理取10mL玻璃离心管,加0.9μg/mL内标溶液20μL,加入1mL人的尿样,涡旋混匀30s,加4mL乙酸乙酯,涡旋振荡3min,3500rpm离心10min,上清液3.5mL于37℃水浴中氮气流挥干,用100μL流动相复溶残渣,22000rpm离心10min一次,取上清液10μL进样分析。2.2UPLC-MS/MS分析条件2.2.1色谱条件色谱柱:ThermoBDSHYPERSILC18100*4.6(mm),2.4μm;流动相:0.3mmol/L醋酸铵(pH6.8)溶液:甲醇=74:26;柱温:35℃;流速:0.4mL/min;进样量:10μL。2.2.2质谱条件ESI离子源,正离子选择性反应检测;定量分析的离子分别为:m/z266.0→166.0(PNA)、m/z252.0→152.0(DDG)、m/z166.0→149.0(O6-甲基鸟嘌呤)、m/z248.0→202.0(替硝唑);碰撞能量分别为:5V(PNA)、5V(DDG)、17V(O6-甲基鸟嘌呤)、10V(替硝唑);鞘气流速:35L/min;雾化气流速:15L/min,毛细管温度:350℃;蒸发气温度:300℃;喷雾电压:3500V。3方法确证3.1专属性空白尿样、空白尿样中加入PNA(3000ng/mL)、DDG(3000ng/mL)、O6-甲基鸟嘌呤(6000ng/mL)和内标替硝唑(18ng/mL)、尿样样品的色谱图分别见图4A、4B和4C,从图中可以看出,空白尿样中的内源性物质不干扰PNA、DDG、O6-甲基鸟嘌呤和内标的测定,内标替硝唑、PNA、DDG、O6-甲基鸟嘌呤的保留时间分别为3.7min、5.1min、2.5min和5.2min。3.2标准曲线PNA、DDG、O6-甲基鸟嘌呤的标准系列溶液由甲醇配制。取空白尿样1mL,加入内标溶液20μL,再依次加入PNA、DDG、O6-甲基鸟嘌呤标准系列溶液,配制成相当于PNA浓度为0.1、0.2、0.5、1、3、10、30、100、300、1000、3000ng/mL,DDG浓度为0.1、0.2、0.5、1、3、10、30、100、300、1000、3000ng/mL,O6-甲基鸟嘌呤浓度为0.2、0.4、1、2、6、20、60、200、600、2000、6000ng/mL的血浆样品,按2.1项下操作,进行UPLC-MS/MS分析,用加权(W=1/x2)最小二乘法进行回归运算,根据标准曲线,本方法PNA的线性范围为0.1~3000ng/mL,标准曲线的方程为:y=0.1661x-0.02617(r=0.9967),DDG的线性范围为0.1~3000ng/mL,标准曲线的方程为:y=0.0009944x-0.001352(r=0.9996),O6-甲基鸟嘌呤的线性范围为0.2~6000ng/mL,标准曲线的方程为:y=0.07229x-0.1748(r=0.9962),y表示待测物与内标的峰面积之比,x表示待测物的浓度。3.3提取回收率取空白尿样1mL,按3.2项下操作,制备低、中、高三个浓度(PNA0.2、10、1000ng/mL;DDG0.2、10、1000ng/mL、O6-甲基鸟嘌呤0.4、20、2000ng/mL)的质控样品,每个浓度进行5样本分析,记录色谱峰。同时,分别取与低、中、高三种浓度水平时等量的标准液,置10mL具塞离心管中,每种水平样品各2份,各精密加入内标溶液20μL(0.9μg/mL),于37℃水浴中氮气流吹干,残留物以100μLUPLC-MS/MS测定用流动相涡旋溶解,22000rpm离心10min。吸取上清液10μL进样分析,获得相应峰面积,以二者色谱图的峰面积之比,考察样品的提取回收率。测定和分析结果见表5-1~5-3。表5-1PNA的提取回收率(n=5)表5-2DDG的提取回收率(n=5)表5-3O6-甲基鸟嘌呤的提取回收率(n=5)如表5-1~5-3所示,PNA三个浓度的提取回收率分别为70.9%、71.7%、71.4%。DDG的提取回收率分别为69.8%、70.4%、74.8%。O6-甲基鸟嘌呤的提取回收率分别为69.2%、70.7%、72.6%。3.4精密度与准确度按3.2项下操作,制备PNA、DDG、O6-甲基鸟嘌呤低、中、高三个浓度的质量控制样品,每个浓度进行5样本分析,连续测定三天,方法精密度由求得的日内、日间RSD(%)来评价,准确度由实际测得值与理论值的偏差来评价,分析结果如表6-1~6-3所示。表6-1尿样样品中PNAUPLC-MS/MS测定方法的准确度与精密度(每浓度每天5样品,连续3天)表6-2尿样样品中DDGUPLC-MS/MS测定方法的准确度与精密度(每浓度每天5样品,连续3天)表6-3尿样样品中O6-甲基鸟嘌呤UPLC-MS/MS测定方法的准确度与精密度(每浓度每天5样品,连续3天)如表6-1~6-3所示,PNA日内、日间精密度分别在7.6%及8.1%以内,准确度范围为91.5~100.7%;DDG日内、日间精密度分别在8.3%及9.4%以内,准确度范围为96.5~103.1%;O6-甲基鸟嘌呤日内、日间精密度分别在7.5%及8.7%以内,准确度范围为89.1~104.3%。表明本方法具有良好的精密度与准确度。3.5稳定性以-20℃冷冻10天及冷冻-溶化三次来考察PNA、DDG、O6-甲基鸟嘌呤的稳定性,如果实测值与理论值的偏差在±15%以内,则表明样品稳定,本实验方法的稳定性结果见表7-1~7-3。表7-1PNA稳定性结果(n=3)表7-2DDG稳定性结果(n=3)表7-3O6-甲基鸟嘌呤稳定性结果(n=3)如表7-1~7-3所示,PNA、DDG、O6-甲基鸟嘌呤浓度的实测值与理论值的偏差在±15%以内,表明本方法具有良好的稳定性。3.6基质效应取空白尿样1mL共15份(5种不同人的来源、每种三份),置10mL玻璃离心管中,加入乙酸乙酯5mL,涡旋振荡3min。3500rpm离心10min,取上清液4mL于另一玻璃离心管,分别加入PNA、DDG、O6-甲基鸟嘌呤标准液适量,配成低、中、高三个浓度水平,每种水平样品各5份,各精密加入内标溶液20μL(0.9μg/mL),涡旋混匀后37℃水浴中氮气流吹干,用100μL流动相复溶残渣,22000rpm离心10min一次,取上清液10μL进样分析。另分别取与基质样品组低、中、高三个浓度水平时等量的PNA、DDG、O6-甲基鸟嘌呤标准液,置10mL具塞离心管中,每种水平样品各5份,各精密加入内标溶液20μL(0.9μg/mL),于37℃水浴中氮气流吹干,用100μL流动相复溶残渣,22000rpm离心10min一次,取上清液10μL进样分析。结果表明基质对两者离子化效率的影响基本稳定(表8-1~8-3)。表8-1PNA基质效应结果(n=5)浓度(ng/mL)基质效应(%)RSD(%)0.289.1±8.59.51096.4±7.17.41000105.2±5.95.6表8-2DDG基质效应结果(n=5)浓度(ng/mL)基质效应(%)RSD(%)0.2100.3±8.48.41095.1±5.96.21000101.7±5.85.7表8-3O6-甲基鸟嘌呤基质效应结果(n=5)
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1