用在凝固测定中的鞣花酸制剂的制作方法
优先权声明本申请要求2014年9月26日提交的美国临时申请号62/055,768的优先权,其整个内容通过引用并入本文。本发明涉及鞣花酸制剂,且具体地,涉及在试验装置中使用鞣花酸制剂进行凝固测定。
背景技术:
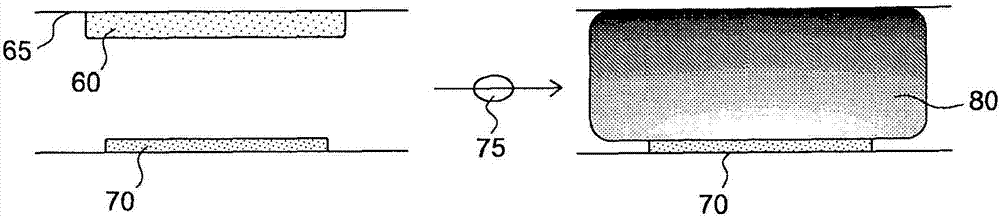
:血液凝固或止血是身体的一种重要保护机制,用于封闭由对身体的损伤造成的创伤。止血以两个阶段发生。初步(细胞的)止血用于快速地停止出血和使失血最小化。初步止血涉及内皮的受损细胞和下面的细胞层,其发出信号使血小板(凝血细胞)能够积累在受损血管区域中,从而形成暂时封闭创伤的栓。次级(血浆的)止血或凝固与初步止血同时开始,并涉及使血液凝固的过程。更具体地,凝固由信号传递凝固级联控制,所述信号传递凝固级联由13种相互作用并相互活化的凝固因子组成。在凝固级联结束时,纤维蛋白原被转化成纤维蛋白。纤维蛋白纤维的网络会补强创伤闭合,且血小板和其它血细胞被捕获在该网络中并形成血块(血栓)。最后,血小板和内皮释放生长因子控制创伤-愈合过程。在这些过程结束时,纤维蛋白网络被血浆中的酶溶解。次级止血的凝固级联是基于纤维蛋白原(一种可溶性血浆蛋白)向不溶性纤维蛋白的催化转化。催化该反应的酶是凝血酶,其不会永久地以活性形式在血液中循环,但是作为凝血酶原(凝血酶的无活性前体)存在。导致有活性的凝血酶的凝固级联由两个途径组成:外源性(extrinsic)途径和内源性(intrinsic)途径,它们汇合成一个共同途径,该共同途径包括催化纤维蛋白原向纤维蛋白的转化的活性凝血酶。外源性途径响应于组织因子(因子iii)的释放而在损伤部位处开始,且因而也被称作组织因子途径。组织因子是因子viia催化的因子x(无活性的)向因子xa(有活性的)的活化中的辅因子。第二个更复杂的内源性途径被与血小板有关的凝固因子viii、ix、x、xi和xii活化。还需要蛋白前激肽释放酶(pk)和高分子量激肽原(hk或hmwk)、以及从血小板分泌的钙离子和磷脂。这些组分中的每一种导致因子x向因子xa的转化。两个途径中的共同点是因子x向因子xa的活化。因子xa是在两个位置(arg-thr,然后arg-ile键)切割凝血酶原的酶(例如,丝氨酸内肽酶),这会产生活性凝血酶并最终导致纤维蛋白原向纤维蛋白的转化。结果,所述凝固级联是用于诊断和治疗涉及失调的血液凝固或凝固缺失的疾病的合适靶标。例如,通过多种凝固试验可以实现出血性病症诸如血友病(其中在凝固级联中涉及的13种血液凝固因子中的一种或多种可能是有缺陷的)的诊断。另外,已经开发了几个试验来监测溶血栓疗法的进展。已经开发了其它试验来发出血栓溶前或高凝固状态的信号或者监测在心肺旁路手术过程中给患者施用鱼精蛋白的影响。但是,凝固试验的主要价值是在监测口服和静脉内抗凝固疗法中。三个关键诊断试验是凝血酶原时间(pt)、激活部分促凝血酶原激酶时间(activatedpartialthromboplastintime)(aptt)和激活凝固时间(act)。已知通过外源性途径和内源性途径中的每一种实现的血液凝固的活化剂。例如,内源性途径的活化可以通过多种带负电荷的不溶物发生。这些物质的作用涉及因子xii和接触活化系统的其它蛋白的特异性吸附,从而导致因子xii、前激肽释放酶和因子xi的蛋白水解性活化的加速。尽管大多数已知的内源性途径的活化剂诸如玻璃、硅石(silica)、c盐(celite)和高岭土是不溶性的,但是已经报道活化通过聚阴离子肝素、硫酸葡聚糖、角叉菜胶和可溶性的鞣花酸而发生(参见,例如,girolami等人,1966,blood,27(1):93-102)。在这些物质中独特的是鞣花酸,它是一种在许多水果、坚果和种子中发现的普遍存在的多酚,和一种有效的活化的cephaloplastin试剂(商购可得的cephaloplastin试剂通常也包括磷脂(通常是脑磷脂)作为血小板替代物)。不幸的是,鞣花酸的水不溶解性不支持它在许多凝固测定(诸如被设计成用在一次性使用的凝固测定装置中的aptt)中作为活化剂的应用,因为包含鞣花酸的活化剂经常需要长时间来制备且在长期储存(例如,在室温大于1-2天)以后是不稳定的。为了解决鞣花酸的水不溶解性,已经提出了许多方案。例如,已经证实鞣花酸可溶于非质子的极性溶剂诸如n,n-二甲基甲酰胺、γ丁内酯、乙腈和n-甲基-2-吡咯烷酮(nmp)(参见,例如,reitze等人,2001,holzforschung,55:171-5)。但是,这些溶剂要么是有毒的,不与凝固测定相容,要么不适合用在一次性使用的凝固测定装置中,因为它们造成安全性、储存期限和其它问题。可替换地,通过添加氢氧化钠已经制备了鞣花酸的可溶性盐(参见,例如,美国专利号3,486,981)。但是,可溶性的盐制品会产生鞣花酸的衍生物,未溶解的鞣花酸,从而使得它们在一次性使用的凝固测定装置中的应用不太合乎需要。另外,含有氢氧化钠的鞣花酸制品倾向于对于较长的时间段不稳定,且因而,通常将防腐剂诸如苯酚加入鞣花酸制品中以使溶液稳定较长的时间段(参见,例如,美国专利号5,055,412)。但是,这些溶液经常需要长时间来制备,这是一个会增加费用的制备缺点。另外,苯酚是腐蚀性的并造成肝和肾损伤,是一种诱变剂和潜在的生殖危害。结果,上述溶解鞣花酸的方案中的许多不适合用于制备作为一次性使用的凝固测定装置中的试剂的鞣花酸。例如,许多有机溶剂是有毒的,且不可用于长期室温储存,后者是大多数一次性使用的(singleuse)一次性(disposable)筒所要求的。此外,为了溶解和稳定鞣花酸而加入的氢氧化物和/或防腐剂会基于醇基的脱水和/或在鞣花酸中存在的两个酯基的皂化反应而产生分解产物(即,从鞣花酸衍生出的无活性化学物质)。因此,需要用于用在一次性使用的一次性筒的血液-凝固测定中的高可溶性的、稳定的和可制备的鞣花酸悬浮液。技术实现要素:在一个实施方案中,本发明涉及一种凝固试验系统,其包含:试剂(reagent),包括鞣花酸和用于稳定地溶解所述鞣花酸的介质(agent),和底物(包括具有可检测部分的、凝血酶可切割的肽)。所述介质包含聚醚化合物和环糊精中的至少一种。在某些实施方案中,所述聚醚化合物选自聚乙二醇、聚环氧乙烷(polyethyleneoxide)、聚氧乙烯(polyoxyethylene)、及其混合物。任选地,所述介质是具有约200至约500000的分子量的聚乙二醇。在某些实施方案中,所述环糊精选自(2-羟丙基)-β-环糊精、(2-羟丙基)-γ-环糊精、α-环糊精、γ-环糊精、二甲基-α-环糊精、三甲基-α-环糊精、二甲基-β-环糊精、三甲基-β-环糊精、二甲基-γ-环糊精、三甲基-γ-环糊精、糖基-α-环糊精、糖基-β-环糊精、二糖基-β-环糊精、麦芽糖基-β-环糊精、二麦芽糖基-β-环糊精、磺丁基醚-β-环糊精、及其混合物。在某些实施方案中,所述介质是环糊精,且所述环糊精与鞣花酸络合。在其它实施方案中,所述介质是聚醚化合物和环糊精,且所述环糊精与鞣花酸络合。任选地,至少80%的鞣花酸是活性鞣花酸。在某些实施方案中,所述凝固试验系统还包含至少一个用聚合物层包被的转换器。所述聚合物层包含具有可检测部分的凝血酶可切割的肽。任选地,所述试剂是激活部分促凝血酶原激酶时间(aptt)试剂,其形成为(i)在所述聚合物层上面的层,或(ii)靠近所述至少一个转换器的层。可替换地,所述试剂是激活部分促凝血酶原激酶时间(aptt)试剂,且所述聚合物层包含所述aptt试剂。在另一个实施方案中,本发明涉及一种形成凝固试验系统的方法,所述方法包括:制备活化剂溶液,其包含鞣花酸和用于稳定地溶解所述鞣花酸的介质,所述介质包含聚醚化合物和环糊精中的至少一种;将所述活化剂溶液与至少一种磷脂制品混合以产生试剂溶液;和将所述试剂溶液加入包含具有可检测部分的、凝血酶可切割的肽的底物以产生测定混合液(cocktail)。在某些实施方案中,所述方法还包括:制备包含聚合物的聚合物基质;和将所述聚合物基质加入所述测定混合液中;和将所述测定混合液分配在芯片的至少一个转换器上。任选地,所述聚醚化合物选自聚乙二醇、聚环氧乙烷、聚氧乙烯、及其混合物。在某些实施方案中,所述介质是聚乙二醇。在其它实施方案中,所述介质是环糊精,且所述环糊精与鞣花酸络合。在替代实施方案中,所述介质是聚醚化合物和环糊精,且所述环糊精与鞣花酸络合。在某些实施方案中,制备所述活化剂溶液包括:在中性ph将鞣花酸溶解在所述含有介质的活化剂溶液中,将碱加入所述活化剂溶液以将所述活化剂溶液的ph增加至10-12.5的ph范围,将所述活化剂溶液混合约1-10分钟的时间段,将酸加入所述活化剂溶液以将ph恢复至中性ph从而保留至少80%的鞣花酸作为活性鞣花酸,和将所述活化剂溶液与缓冲液混合。在另一个实施方案中,本发明涉及一种样品分析筒(cartridge),其包含:被构造成容纳生物样品的入口室;导管,其与所述入口室流体连通且被构造成容纳来自所述入口室的生物样品;和位于所述导管内的微环境激活部分促凝血酶原激酶时间(aptt)传感器,所述传感器包含含有鞣花酸和用于稳定地溶解所述鞣花酸的介质的试剂以及至少一个用聚合物层包被的转换器,其中所述聚合物层包含具有可检测部分的、凝血酶可切割的肽。附图说明考虑到以下非限制性附图将更好地理解本发明,在以下非限制性附图中:图1显示了根据本发明的某些方面的筒示意图;图2和3显示了根据本发明的某些方面的包含可溶解性试剂/底物和转换器的导管;图4显示了根据本发明的某些方面的可扩散性试剂、固定化的底物-聚合物层和转换器;图5和6显示的图提供了本发明的方面的实验证据;图7a、7b和7c图解了根据本发明的某些方面的微环境传感器的运行原理,所述微环境传感器包含试剂和/或底物(固定化在聚合物层中或不在聚合物层中)和转换器;图8显示了根据本发明的某些方面的筒示意图;图9显示了根据本发明的某些方面的固定化的试剂/底物-聚合物层的制造的侧视图;图10-12显示的图提供了本发明的方面的实验证据;图13a、13b和13c显示了根据本发明的某些方面的可扩散性试剂、固定化的底物-聚合物层和转换器的多种布置(arrangement);图14显示了根据本发明的某些方面的传感器的制造的侧视图;图15和16显示了根据本发明的某些方面的多种传感器构型;图17显示了根据本发明的某些方面的一次性传感装置的顶视图;图18-20显示了根据本发明的某些方面的多种传感器构型;图21、22a和22b图解了根据本发明的某些方面的电导测定传感器的运行原理;图23显示了根据本发明的某些方面的一次性传感装置和读出装置的等轴视图;图24显示了根据本发明的某些方面的一次性传感装置的顶视图;图25和26显示了根据本发明的某些方面的一次性传感装置的一部分的顶视图;图27-29显示了根据本发明的某些方面的高级微流体系统;图30显示了根据本发明的方面的独立混合控制的图;图31显示了根据本发明的某些方面的一次性传感装置的一部分的顶视图;图32和33显示了根据本发明的某些方面的高级微流体系统;图34显示了根据本发明的某些方面的一次性传感装置的一部分的顶视图;图35显示了根据本发明的某些方面的高级微流体系统;图36显示了根据本发明的某些方面的一次性传感装置的一部分的顶视图;图37和38显示了根据本发明的某些方面的高级微流体系统;图39显示了根据本发明的某些方面的一次性传感装置的一部分的顶视图;图40、41a和41b显示的图提供了本发明的方面的实验证据;图42和43图解了根据本发明的某些方面消除接地芯片的运行原理;图44显示了在氢氧化钠中新鲜制备的鞣花酸制品的hplc色谱图;图45显示了在氢氧化钠中制备并在室温储存2天的鞣花酸制品的hplc色谱图;图46显示了在甲醇中新鲜制备的鞣花酸制品的hplc色谱图;图47显示了在甲醇中制备并在室温储存6天的鞣花酸制品的hplc色谱图;图48显示了在甲醇中制备并在室温储存13天的鞣花酸制品的hplc色谱图;图49显示了在1mpeg400中制备并在水中稀释的新鲜鞣花酸制品的hplc色谱图;图50显示了在1mpeg400中制备、在室温储存6天并在甲醇中稀释的鞣花酸制品的hplc色谱图;图51显示了在1mpeg400中制备、在室温储存13天并在甲醇中稀释的鞣花酸制品的hplc色谱图;图52显示了在100mmpeg4000中制备并在室温储存6天的鞣花酸制品的hplc色谱图;图53显示了在100mmpeg4000中制备并在室温储存13天的鞣花酸制品的hplc色谱图;图54显示了用β-环糊精制备的新鲜鞣花酸制品的hplc色谱图;图55显示了用β-环糊精制备并在室温储存6天的鞣花酸制品的hplc色谱图;图56显示了用ph休克处理的β-环糊精制备的新鲜鞣花酸制品的hplc色谱图;图57显示了用ph休克处理的(shocked)β-环糊精制备并在室温储存6天的鞣花酸制品的hplc色谱图;图58显示了使用aptt测定试剂时电流相对于时间的计时电流测定图;和图59显示了使用aptt测定试剂时电流相对于时间的计时电流测定图。发明详述本发明涉及鞣花酸制剂,且具体地,涉及在试验装置中使用鞣花酸制剂进行凝固测定。在优选的实施方案中,本发明涉及用在凝固测定中的组合物和制备高可溶形式的鞣花酸(例如,凝固活化剂)的方法。所述鞣花酸可以以这样的形式制备:其包括在凝固测定中变得迅速溶解的干燥试剂。额外地或可替换地,所述鞣花酸可以以包括处于微环境形式的聚合物层的形式制备,以便物理上分离由鞣花酸的存在产生的反应性组分,从而避免交叉活化和促进电化学或光信号在至少一个转换器上面的定位。在某些实施方案中,所述鞣花酸可以用于促进aptt测定中的内源性途径的活化。在某些实施方案中,本发明涉及一种组合物和制备凝固测定试剂的方法,所述方法包括:将鞣花酸溶解在氢氧化钠、甲醇、聚醚化合物(特别是聚乙二醇、聚环氧乙烷或聚氧乙烯)和环糊精客体-主体复合物中的一种或多种中。为了在使鞣花酸中的醇基脱水和/或酯键皂化最小化的同时促进溶解,所述组合物和制备凝固测定试剂的方法还可以包含将鞣花酸暴露于短暂的碱性ph漂移(excursion)。在某些实施方案中,所述凝固测定试剂可以用于促进aptt测定中的内源性途径的活化。根据本文中公开的本发明的这些和其它方面,所述组合物和制备鞣花酸和凝固测定试剂的方法有利地获得这样的鞣花酸制剂和凝固测定试剂:其对于长期储存而言是非常稳定的,并减少aptt凝固测定的测定时间。本文中使用的术语“微环境传感器”表示这样的传感器:其被构造成使得以足够在传感器处实现期望的信号的方式在所述传感器紧邻处发生的任何反应不会可检测地干扰(或影响)在正常使用过程中在邻近传感器处发生的另一个反应。本文中使用的术语“中和肝素”表示传感器使生物样品中的未分级分离的肝素和低分子量肝素(lmwh)在足以跨微环境传感器区域的区域中无生物活性的方面。相反,“不中和肝素”表示传感器不会影响(impact/affect)微环境传感器区域中的未分级分离的肝素或lmwh的生物活性的方面。本文中使用的术语“固定化”表示微环境传感器在运动方面基本上受限的方面,且因而将微环境的该方面定位至一般区域。本文中使用的术语“底物”表示为酶反应的靶标的分子或形成结构的基础的物理实体。血液凝固的概述血液凝固过程和随后在修复受损组织以后凝块的溶解被称作止血。为了使止血发生,血小板必须附着于暴露的胶原,释放它们的颗粒的内容物,并聚集。血小板向在内皮细胞表面上暴露的胶原的附着由vonwillebrand因子(vwf)介导。经由凝血酶对血小板的活化是它们随后聚集成血小板栓所必需的。但是,同样重要的是活化的血小板表面磷脂在凝固级联的活化中的作用。凝固级联的内源性途径需要凝固因子viii、ix、x、xi和xii。还需要蛋白前激肽释放酶(pk)和高分子量激肽原(hk或hmwk)、以及从血小板分泌的钙离子和磷脂。这些内源性途径组分中的每一种导致因子x向因子xa的转化。当前激肽释放酶、高分子量激肽原、因子xi和因子xii暴露于带负电荷的表面时,发生内源性途径的起始。这被称作接触阶段且可以作为与循环脂蛋白颗粒(诸如乳糜微粒)的磷脂(主要是磷脂酰乙醇胺,pe)、极低密度脂蛋白(vldl)和氧化的低密度脂蛋白(ldl)的相互作用的结果而发生。这是高脂血症在前血栓状态的促进中的作用的基础。因子xa在内源性途径中的活化要求tenase复合物(ca2+和因子viiia、ixa和x)在活化的血小板的表面上组装。血小板对活化的应答之一是磷脂酰丝氨酸(ps)和磷脂酰肌醇(pi)在它们的表面上的呈现。这些磷脂的暴露允许tenase复合物形成并随后活化因子xa。凝固级联的外源性途径在损伤部位处响应于组织因子(因子iii)的释放而开始,且因而也被称作组织因子途径。组织因子是因子viia催化的因子x活化中的辅因子。因子viia(含有gla残基的丝氨酸蛋白酶)以与内源性途径的因子ixa相同的方式将因子x切割成因子xa。因子vii的活化通过凝血酶或因子xa的作用而发生。因子xa的活化因子vii的能力会建立内源性途径和外源性途径之间的关联。两个途径中的共同点是因子x活化为因子xa。因子xa将凝血酶原(因子ii)活化为凝血酶(因子iia)。凝血酶又将纤维蛋白原转化成纤维蛋白。凝血酶的活化发生在活化的血小板的表面上,且需要凝血酶原酶复合物的形成。该复合物由血小板磷脂、磷脂酰肌醇和磷脂酰丝氨酸、ca2+、因子va和xa以及凝血酶原组成。因子v是在凝血酶原酶复合物的形成中的辅因子,类似于因子viii在tenase复合物形成中的作用。象因子viii活化一样,因子v借助于微小量而活化为因子va,且被增加水平的凝血酶灭活。因子va结合活化的血小板的表面上的特定受体且与凝血酶原和因子xa形成复合物。凝血酶原是一种在它的n-端区域中含有10个gla残基的72kda单链蛋白。在凝血酶原酶复合物内,凝血酶原在2个位点处被因子xa切割。该切割产生含有a和b链的2-链活性凝血酶分子,所述a和b链通过单个二硫键保持在一起。凝血酶结合一类g-蛋白偶联受体(gpcr),其被称作蛋白酶活化的受体(par),特别是par-1、-3和-4。par利用一种独特机制将细胞外蛋白水解性裂解的结果转化成细胞内信号传递事件。par携带它们自己的配体,其在蛋白酶切割(诸如被凝血酶切割)“暴露”所述配体之前保持无活性。在凝血酶切割之后,被暴露的配体仍然是完整par的一部分,但是现在能够与par的配体结合结构域相互作用,从而导致众多信号传递级联的活化。凝固试验的概述使用出血时间测定来评价与止血有关的血管和血小板应答。出血时间是在外科手术之前在外科手术前患者上进行的常见测定,以确保存在对血管损伤的适当应答。如本文所讨论的,对血管损伤的快速应答(在几秒内发生)是血管收缩和血小板向血管壁的附着。用于确定出血时间的ivy方法包括血压袖袋(血压计)的应用,所述血压袖袋被放置在前臂上且膨胀至40mmhg。然后在前臂上产生一个浅表切口并记录出血停止所需的时间。对于ivy方法,出血应当在1-9分钟内停止。大于15分钟的任何出血时间将指示血管和血小板对血管损伤的初步应答的缺陷。较低侵袭性的出血时间测定包括刺血针或特殊针的应用,用它们在指尖或耳垂上做出3-4mm深的刺扎。该出血时间测定被称作duke方法,且在该测定中出血应当在1-3分钟内停止。出血时间受血小板功能的任何缺陷影响(延长),且在血管性血友病中受血管障碍影响,但是不受其它凝固因子影响。通常与增加的出血时间有关的障碍包括血小板减少症、弥散性血管内凝血(dic)、bernard-soulier综合征和glanzmann血小板机能不全。异常的出血时间还见于具有库欣综合征、严重肝病、白血病和骨髓衰竭的患者中。还可以用特定测定来评估与血液凝固途径的因子有关的缺陷。凝血酶原时间(pt)是被设计成筛选纤维蛋白原、凝血酶原和因子ii、v、vii和x的缺陷的测定,且因而测量凝固的外源性途径的活性。当这些因子中的任一种有缺陷时,那么pt被延长。正常的pt是11.0-12.5秒。大于20秒的pt指示凝固缺乏。通常使用除去血细胞以后的血浆测量pt。通常将血液样品收集在含有柠檬酸盐的试管中以结合任何钙并从而抑制凝固,然后将细胞通过离心进行分离。将过量的钙加入血浆的等分试样中以开始凝固。最常见的pt的量度是将患者的血液的凝固时间除以平均正常pt值,随后将该比率升高至与正在使用的试剂的isi(国际敏感指数)对应的幂。得到的值被称作国际标准化比率(inr)。正常值的范围为0.8-1.2inr。使用pt来确定香豆素类抗-凝固药物(例如)的正确剂量、肝病或损伤的存在,并评价维生素k状态。使用激活部分促凝血酶原激酶时间(aptt)来测定凝固的内源性途径中的缺陷。aptt测定包括添加活化剂,所述活化剂会缩短正常凝固时间且在具有未解释的出血或凝固的患者中经常开处方。所述测定会评价纤维蛋白原、凝血酶原和因子v、viii、ix、x、xi和xii的功能。这些因子中的任一种的缺陷将导致延长的aptt。正常的aptt是30-40秒。aptt是用于评估肝素抗凝血疗法的效力的标准测定。通常使用除去血细胞以后的血浆测量aptt。通常将血液样品收集在含有柠檬酸盐的试管中以结合任何钙并从而抑制凝固,然后将细胞通过离心进行分离。将过量的钙加入血浆的等分试样中以逆转柠檬酸盐抗凝固。延长的aptt与获得性或先天性出血障碍有关,所述获得性或先天性出血障碍与凝固因子缺乏、维生素k缺乏、肝病、dic、血管性血友病、白血病、血友病和肝素施用有关。激活凝固时间(act)是用于监测高剂量肝素疗法或比伐芦定治疗的常见现场护理(pointofcare)全血凝血试验。在这些场合中需要的肝素或比伐芦定的剂量超出用aptt可以测量的范围。通常,将全血收集进含有凝固活化剂(例如,硅藻土、高岭土或玻璃颗粒)和磁力搅拌棒的试管或筒中,并然后测量血液凝固所用的时间。act的参考值通常在70-180秒之间的范围内。抗凝固的合乎需要的范围取决于适应症和使用的试验方法。例如,在心肺旁路手术过程中,使用肝素的期望act范围可以超过400-500秒。相反,在接受经皮冠状动脉干预的患者中,当与糖蛋白iib/iiia拮抗剂结合施用肝素时,支持200秒的靶act,而在没有这样的辅助治疗存在下靶向250-350秒之间的act。用于确定诊断性凝固时间的电化学系统已经使用产色测定通过对特定因子特异性的人工的可切割的肽底物的发展来测量特定凝固因子的酶活性。应当指出,基于凝固时间(诸如aptt、pt和act)的测定是在有抗凝血剂(诸如华法林和肝素)或有缺陷的凝固因子存在下凝血酶形成和抑制的基本上功能性量度。因而,可以在基于纤维蛋白形成的测量的测定和直接基于凝血酶活性的测量的测定(通过使用适当的肽底物,如在产色测定中)之间做出类比。电化学检测包括工作电极(例如,电流测定电极)和参比电极(例如,计数器参比电极)的应用,其中将恒定电位施加于工作电极从而产生氧化-还原(氧化还原)反应,其可以量化为可记录的电流。电化学传感器已经广泛应用在现场护理(poc)和自-试验装置的开发中,如葡萄糖试验条的开发所例证的,因为它们与电子仪器的接口是简单的且降低了装置成本。装置,诸如系统(参见,例如,美国专利号7,977,106,其整个内容通过引用并入本文),已经采用产电底物,所述产电底物导致与凝血酶活性成比例的可电化学上检测的切割产物的形成。然后将这些装置构造成基于凝血酶活性的量度而返回凝固时间,从而允许与标准凝固进行对比。因此,在某些实施方案中,所述电化学检测系统被称作“产电的”,因为产生可电化学上检测的物质以允许确定速率测量或试验端点,例如,诊断性凝固时间。这类似于产色的或发荧光的端点试验,其中样品的光吸收或发射性能的变化指示速率测量或端点,例如,诊断性凝固时间。图1解释了根据本发明的一些实施方案用于确定诊断性凝固时间的电化学检测系统10(例如,电流测定电化学检测系统)的原理。但是,应当理解,尽管本文关于诊断性凝固时间测定(例如,pt、aptt和act测定)描述了具体实施方案,本文描述的微环境传感器结构也可以用于检测各种可能感兴趣的分析物。更具体地,本发明的电化学检测系统不限于凝固酶的测定。例如,其中酶切割底物分子以产生电活性部分的任何测定可以使用本发明的方法。应当理解,可以为本领域中的多种其它已知酶(例如,葡萄糖氧化酶、乳酸氧化酶和其它氧化还原酶、基于脱氢酶的酶和碱性磷酸酶和其它磷酸酶和丝氨酸蛋白酶)设计测定,而不脱离本发明的范围。例如,本发明的一些方面可能包括磷酸酶测定,其中具有磷酸盐部分的二茂铁存在于微环境传感器层中。存在于样品中的酶磷酸酶可以渗入微环境传感器并切割磷酸酯基团,从而使释放的二茂铁分子能够在电极处被氧化。因此,测量的电流可以随切割反应的速率而变化,且因而与样品中的磷酸酶活性成比例。在一个示例性的分析中,可以将流体样品15(例如,全血)引入本发明的筒25的样品容纳室20中。此后,可以将流体样品15引导至所述筒的分析区域30,例如,传感器区域或所述筒的一个或多个导管内的一个或多个位置,其包括一个或多个传感器,所述传感器用于凝固检测和任选地用于检测靶分析物(例如,凝血酶活性(对于凝血酶原时间)和肌钙蛋白i)。分析区域30包括一个或多个微环境传感器35,其包含处于任意数目的不同可能布置的一个或多个电极或转换器37、一种或多种试剂40和一种或多种底物45。所述电极、试剂和底物的形式和取向可以随本发明的实施方案宽泛地变化,这将在后面详细描述。根据本发明的某些方面,所述一种或多种试剂40可以包括通过内源性或外源性途径诱导凝固的物质。适合用于诱导外源性途径(例如,pt分析)的物质可以包括一种或多种选自以下的组分:非重组组织因子、重组组织因子、合成的或天然的脂质、合成的或天然的磷脂、合成的或天然的脂质的组合和合成的或天然的磷脂的组合。在某些实施方案中,可以在所述一种或多种试剂40内包括多种其它组分以促进所述一种或多种试剂40的稳定和沉积/溶解特征。例如,所述一种或多种试剂40还可以包含一种或多种选自以下的组分:载体蛋白诸如牛血清白蛋白(bsa)、稳定剂、抗微生物剂、钙盐、钾盐、水溶性的聚合物、糖(sugar)、明胶、琼脂糖、多糖、糖化物(saccharide)、蔗糖、聚乙二醇、磷酸钠、甘氨酸、氨基酸、抗氧化剂、去污剂、缓冲盐和缓冲剂诸如4-(2-羟基乙基)-1-哌嗪乙磺酸(hepes)缓冲剂。根据本发明的不同方面,所述一种或多种试剂40可以包括适合用于诱导内源性途径的物质。适合用于诱导内源性途径(例如,aptt或act分析)的物质可以包括一种或多种选自以下的组分:鞣花酸、硅藻土、高岭土、硅藻土、粘土、二氧化硅、合成的或天然的脂质、和合成的或天然的磷脂。在某些实施方案中,可以在所述一种或多种试剂40内包括多种其它组分以促进所述一种或多种试剂40的稳定和/或沉积/溶解特征。例如,所述一种或多种试剂40还可以包含一种或多种选自以下的组分:葡聚糖、环糊精、糊精、tergitol、缓冲剂、载体蛋白、氨基酸、稳定剂、抗微生物剂、抗氧化剂、去污剂、糖化物(saccharide)、多糖、蔗糖、聚醚化合物诸如聚乙二醇、聚乙二醇衍生物、聚环氧乙烷或聚氧乙烯、甘氨酸、明胶、缓冲剂诸如4-(2-羟基乙基)-1-哌嗪乙磺酸(hepes)缓冲剂、鼠李糖、海藻糖和糖。根据本发明的某些方面,在产电测定中使用的一种或多种底物45可以具有酰胺键,其模仿凝血酶切割的纤维蛋白原中的酰胺键。具体地,所述一种或多种底物45可以包含一种或多种凝血酶可切割的肽诸如选自以下的那些:h-d-phe-pip-arg、h-d-chg-abu-arg、cbz-gly-pro-arg、boc-val-pro-arg、h-d-phe-pro-arg、环己基甘氨酸-ala-arg、tos-gly-pro-arg、bz-phe-val-arg、boc-val-pro-arg、ac-val-pro-arg、ac-val-hyp-arg、ac-(8-氨基-3,6-二氧杂辛酰基-val-pro-arg、ac-gly-pro-arg、ac-(8-氨基-3,6-二氧杂辛酰基-gly-pro-arg、ac-gly-hyp-arg和h-d-chg-abu-arg。凝血酶通常在精氨酸残基的羧基端处切割酰胺键,因为所述键在结构上类似于凝血酶切割的纤维蛋白原中的酰胺键。凝血酶-底物反应的产物包括电化学上惰性的化合物诸如tos-gly-pro-arg、h-d-phe-pip-arg和/或bz-phe-val-arg-和电活性的化合物或可检测的部分,其优选地选自对-氨基苯酚、醌、二茂铁、亚铁氰化物衍生物、其它有机金属物质、对-硝基苯胺、邻-二茴香胺、4,4’-联苯胺、4-甲氧基-2-萘基胺、n-苯基-对苯二胺、n-[对甲氧基苯基-]-对苯二胺和吩嗪衍生物。选择三肽序列,因为它使得所述底物基本上不与除了凝血酶以外的血液蛋白酶反应,且凝血酶与分子中的精氨酸酰胺键的反应性非常类似于它与纤维蛋白原中的靶酰胺键的反应性。当一种或多种底物45存在于血液或血液衍生物流体样品或生物样品中时,从所述一种或多种试剂40对凝固途径的活化产生的活性凝血酶同时将所述一种或多种底物45和纤维蛋白原转化成它们的切割产物。通过所述一个或多个转换器37(例如,电化学转换器)来检测电化学物质反应产物。微环境传感器结构如本文所讨论的,微环境传感器结构包含处于许多不同布置中的任一种的一种或多种试剂和一种或多种底物,使得流体样品(例如,全血)向所述一种或多种试剂和所述一种或多种底物的引入被定位至所述一个或多个传感器。具体地,所述微环境传感器结构被构造成将一种或多种试剂和/或反应产物彼此物理上分离以在一种或多种试剂已经变得暴露于流体样品以后避免级联途径的交叉活化或其它传感器交叉干扰。如在图2中所示,传统的poc凝固测定已经采用试剂/底物60,所述试剂/底物60作为干燥物质印制在与传感器70的表面相对的导管的壁65(例如,盖子)上。需要将流体样品75与干燥物质混合(例如,通过泵振荡),以将试剂/底物60溶解在流体样品75中和产生混合物80,其可以呈从导管顶部向下至传感器70的梯度的形式。但是,这样的构型具有至少三个问题或缺点。首先,小部分由混合物80产生的电活性产物到达传感器70的表面并被氧化,且因而大部分电活性产物不会被利用。所以,试剂/底物60的使用不是有效的。进一步,流体样品75掺杂了试剂/底物60,这可能是不希望的,因为可能影响可与流体样品75发生接触的其它传感器(例如,传感器交叉干扰)。其次,为了达到适当的分析精确度,应当尽可能快速地将试剂/底物60均匀地分散在流体样品75中。这对于其中空间和混合效率可能有限的现场护理装置而言可能是一个挑战。当试剂/底物60呈固体形式和与流体样品75的体积相比具有非常小的空间时,这是特别真实的。第三,存在底物干扰试剂和/或凝固因子的可能性。例如,在凝固级联已经开始之前将底物与试剂一起混合进样品75中可能表现出这样的干扰。与传统的poc凝固测定不同,本发明的一些实施方案(如在图3中所示)提出与底物层90结合的试剂85,所述底物层90以定位方式形成在传感器95的表面附近。例如,如在图3中所示,试剂85和底物90可以作为干燥物质直接印制在传感器95的表面上。流体样品100可以在没有混合(例如,通过被动扩散)的情况下以定位方式与试剂85和底物90反应(尽管可能期望某种程度的混合,例如,流体振荡),从而建立从传感器95至导管顶部的梯度。有利地,直接呈现在传感器表面上的试剂和底物的这种布置允许大部分电活性产物被氧化,并因而在传感器的表面处被利用。由于在紧邻传感器环境中需要较小的样品体积,这种传感器布置也是有益的,并因而产生更浓的试剂-至-样品测定地带。尽管如此,图3中所示的布置没有克服在传统的poc凝固测定中显而易见的一些问题(例如,传感器交叉干扰和底物干扰的减轻)。例如,在传感器的邻近附近中发生的任何反应潜在地干扰试剂和/或凝固因子和/或可能干扰在靠近的传感器(即,在相同导管内和在图3所示的传感器的大约3mm内的传感器)处发生的另一个反应。这样,这类传感器布置不会被表征为微环境传感器。但是,这些剩余的问题可以通过如下文详细讨论的本发明的高级微流体系统(例如,将单个样品分成两个或更多个部分和控制那些部分向两个或更多个导管中的移动)和/或传感器彼此的适当间隔来克服。例如,在某些实施方案中,在靠近的传感器被相同静止样品流体覆盖的情况下,为了防止低于给定阈值(例如,低于1%)的传感器交叉干扰,可能合适的是使用基于干扰物的已知扩散系数和总测定时间的模型来确定传感器之间的适当分离距离。在其它实施方案中,在样品不静止的情况下,关于动态混合的其它模型可能适合用于选择适当的传感器分离距离。在额外或替代实施方案中,已经意外地证实将底物90固定化在传感器95上会解决许多或全部上述问题。根据本发明的这些方面,通过交联(例如,紫外线、戊二醛等)、捕集、共价结合等可以实现固定化。这样的微环境布置的一个例子显示在图4中,其中使用聚合物层105将底物90固定化在传感器95的表面上。在某些实施方案中,所述固定化可以如下完成:用包括底物90的聚合物层105涂布传感器95,使得底物90经由聚合物层105固定化在传感器95的表面上。换而言之,底物90作为固定化的多孔的底物-聚合物层形成在传感器95的表面上,以在传感器95的表面上以定位方式建立用于维持流体样品100、试剂85和底物90的反应的容器。流体样品100可以在形成于传感器上的聚合物层105(或者在其上面,并然后扩散进其中)的范围内以定位方式在没有混合的情况下与试剂85和底物90反应(尽管可能期望某种程度的混合,例如,流体振荡)。有利地,直接存在于传感器表面上的固定化底物的这种布置允许大量电活性产物被氧化,并因而在传感器的表面处被利用。甚至更有利地,固定化底物的这种布置会提供这样的微环境:其能够将底物和电活性产物维持在传感器的邻近附近,并因而减轻在正常使用过程中与靠近的传感器的传感器交叉干扰。将底物固定化在传感器上的其它潜在益处包括:通过底物与试剂的分离来减轻底物干扰,减少物质使用,简化硬件和传感器设计,和提高产品稳健性。图5和6提供的实验证据表明,将底物固定化可以显著地增加应答电流和提高分析物检测的精确度。具体地,图5显示了aptt应答曲线,其中x-轴是时间/秒且y-轴是电流/pa。在该实施例中,将底物印制在传感器上,一种用pva固定化(aptt应答曲线106),且另一种没有固定化(aptt应答曲线107)。将aptt试剂掺入全血中。混合约30秒以后,将样品从样品试管抽出,并装入筒中用于试验。固定化底物传感器的电流(aptt应答曲线106)超过30na,而未固定化底物传感器的电流(aptt应答曲线107)仅为约3na。它们的tmid(电流达到它的中点时的时间)的变动系数分别是约1%和2%。该数据指示,固定化底物直接在传感器上的存在能够实现从凝固底物离去基团的立即且集中的氧化还原反应。这又产生更快的凝固时间和更可预测的传感器应答。在图6中显示了另一个实施例,其中x-轴是时间/秒且y-轴是电流/pa。pt应答曲线108代表非固定化底物传感器对对照流体水平2的应答,而pt应答曲线109代表固定化的底物传感器对相同对照流体水平2的应答。关于未固定化底物传感器,底物和试剂都被一起印制在电极上,并在试验过程中与样品一起混合。关于固定化底物传感器,将底物用pva固定化在电极上,并将试剂印制在固定化底物的上面,在试验过程中不存在混合。固定化的传感器与血浆对照一起使用会产生性能的显著改善,使得电流从约4na增加至约9na,且变动系数从约10%降低至约3%。该数据表明凝固试验领域中的显著改进,使得现场护理装置的性能现在可以接近中心实验室仪器的性能(2-3%cv)。如在图7a、7b和7c中所示,本发明的微环境传感器可以具有以许多不同布置定位的试剂110和固定化底物-聚合物层115,所述组分在没有混合的情况下彼此相互作用,尽管某种程度的振荡可能是期望的。例如,如在图7a中所示,试剂110可以定位在固定化底物-聚合物层115内或者被固定化底物-聚合物层115包封(例如,所述试剂掺入在固定化底物-聚合物层内)。如在图7b中所示,试剂110可以覆盖在固定化底物-聚合物层115上面(例如,所述试剂是分布在固定化底物-聚合物层上面的单独层)。如在图7c中所示,试剂110可以基本上位于固定化底物-聚合物层115和传感器120的至少一个转换器附近(例如,所述试剂定位在所述导管内,使得所述试剂贴近底物-聚合物层和/或至少一个转换器或在底物-聚合物层和/或至少一个转换器的相互作用距离内,从而仍然彼此联合地起作用)。本文中使用的相互作用距离是指小于所述传感器的最长尺寸,所述试剂的约束是定位在相同平面内或与传感器相同的导管壁/表面上。本领域技术人员还会明白其它变体,而不脱离本发明的精神和范围,例如试剂110可以形成为在图7b和7c中所示的试剂的组合,或如在图7c中所示具有图7b中所示的试剂110的仅一部分。如在图8中所示,在某些实施方案中,本发明可能涉及分析筒125,其包含被构造成容纳流体样品135的入口室130和导管140,所述导管140与入口室130流体连通且被构造成容纳来自入口室130的流体样品135。导管140可以包含大量微环境传感器,例如,第一微环境传感器145和第二微环境传感器150。第一微环境传感器145可以包含被构造成检测第一诊断性凝固时间的第一试剂155和第一底物160(例如,被固定化在聚合物层内的底物)。例如,第一微环境传感器145可以是pt传感器,其包含第一试剂155和第一底物层160,所述第一试剂155包括一种或多种对触发外源性凝血途径特异性的组分(如本文所讨论的),所述第一底物层160包含如本文中讨论的具有可检测部分的、凝血酶可切割的肽。第二微环境传感器150可以包含被构造成检测第二诊断性凝固时间的第二试剂165和第二底物170(例如,被固定化在聚合物层内的底物)。例如,第二微环境传感器150可以是aptt传感器,其包含第二试剂165和第二底物层170,所述第二试剂165包括一种或多种对触发内源性凝固途径特异性的组分(如本文所讨论的),所述第二底物层170包含具有可检测部分的、凝血酶可切割的肽(例如,被固定化在聚合物层内的试剂和底物)。应当理解,尽管关于pt传感器和aptt传感器讨论了上述的分析筒125,本发明预见到传感器(例如,pt传感器、aptt传感器和act传感器)的各种组合和数目,而不脱离本发明的范围。例如,第一微环境传感器145可以是pt传感器,且第二微环境传感器150可以是aptt传感器或act传感器。在另一个方面,第一微环境传感器145是aptt传感器,且第二微环境传感器150是pt传感器或act传感器。在另一个方面,第一微环境传感器145是act传感器,且第二微环境传感器150可以是aptt传感器或pt传感器。在其它实施方案中,所述微环境传感器之一是pt传感器、aptt传感器或act传感器,且所述传感器中的另一个是用于检测与凝固相关或无关的分析物的传感器。有利地,将本发明的微环境传感器结构构造成物理上分离一种或多种试剂和底物以避免所述一种或多种试剂和底物已经变得暴露于流体样品后级联途径的交叉活化和/或干扰。甚至更有利地,固定化的底物和/或试剂聚合物层向凝固测定中的整合会提供在不需要混合(例如,在导管中振荡样品流体)的情况下或在使混合最小化的同时进行凝固测定的能力,因为凝固活化发生在传感器上的定位且集中的区域中,随后试验反应扩展进固定化的层中,最后导致在转换器处的氧化。固定化底物-聚合物层在优选的实施方案中,为了使一个或多个测定彼此物理上分离以避免交叉活化和促进电化学或光信号在转换器上的定位,可以将固定化的底物和/或试剂-聚合物层选择性地模印在传感器上(例如,包被在转换器或工作电极/光学检测器上)。如在图9中所示,可以通过自旋涂布或通过微分配形成固定化的聚合物层175。更具体地,包含一种或多种试剂和底物和聚合物(诸如可光变形的聚合物(例如,聚乙烯醇(pva)))的水性聚合物基质可以用于将一种或多种底物固定化在转换器180上或附近。还可以在水性基质中包括添加剂,包括、但不限于蛋白诸如bsa、糖或糖醇,诸如蔗糖、山梨醇或甘露醇。对于聚合物化学领域的技术人员而言,某些物质向聚合物层的添加会导致包括、但不限于膨胀反应、扩散系数、分子稳定性、孔隙率、运输、反应动力学等的许多改变。这些改变可以用于根据需要调节微环境传感器应答。根据本发明的某些方面,所述一种或多种底物可以包含一种或多种凝血酶可切割的肽,所述肽选自h-d-phe-pip-arg、h-d-chg-abu-arg、cbz-gly-pro-arg、boc-val-pro-arg、h-d-phe-pro-arg、环己基甘氨酸-ala-arg、tos-gly-pro-arg、bz-phe-val-arg、boc-val-pro-arg、ac-val-pro-arg、ac-val-hyp-arg、ac-(8-氨基-3,6-二氧杂辛酰基-val-pro-arg、ac-gly-pro-arg、ac-(8-氨基-3,6-二氧杂辛酰基-gly-pro-arg、ac-gly-hyp-arg和h-d-chg-abu-arg。任选地,可以混合这些底物中的两种或更多种以得到在固定化的底物和/或试剂聚合物层中期望的凝血酶活性和扩散性质。根据本发明的某些方面,含有底物的聚合物可以包含一种或多种物质,任选地以基质形式存在。例如,用于聚合物的物质可以选自:pva、苯乙烯基吡啶聚乙烯醇(sbq-pva)、琼脂糖、聚丙烯酰胺、聚甲基丙烯酸甲酯、n-甲基吡咯烷酮、聚乙烯吡咯烷酮、聚酰亚胺、形成膜的胶乳、琼脂糖凝胶(sepharose)、聚氨酯或聚氨基甲酸酯(polyurethanes)、丙烯酸酯、甲基丙烯酸酯、聚乙二醇、聚乳酸、聚乳酸-乙醇酸共聚物(poly(lacticco-glycolicaicd))、羟丙基纤维素、纤维素、纤维素衍生物、羟丙基甲基醋酸纤维素琥珀酸酯(hydroxypropylmethylcelluloseacetatesuccinate)、菊糖、果聚糖、果聚糖衍生物、聚乙醇酸、elvace、羧甲基纤维素、聚乳酸和聚乳酸-乙醇酸共聚物。在某些其中用于聚合物的物质包含纤维素(例如,羟丙基纤维素)的实施方案中,还可以在水性基质中包括添加剂诸如塑化剂(例如,柠檬酸三乙酯、乙酰基柠檬酸三乙酯、丙二醇、甘油、三羟甲基丙烷、聚乙二醇、脂肪酸、及其衍生物)和/或交联剂(例如,羧酸、乙二醛和任何可与纤维素的可用羟基反应的树脂)。所述物质的交联也可以影响聚合物层膨胀、渗透性、扩散、反应动力学等以便根据需要调节传感器应答。选择用于聚合物的物质以后,固定化底物和/或试剂的另一个益处包括使用固定化基质作为定位的干扰物中和剂。例如,用于聚合物的物质的选择可以依赖于要使用固定化的聚合物层进行的诊断性凝血试验的类型。例如,已经有利地和意外地发现,交联的或非交联的sbq-pva在固定化的聚合物层中的包含(inclusion)会给固定化的聚合物层赋予肝素中和性质或肝素不敏感性。结果,在其中要使用固定化的聚合物层进行的诊断性凝血试验是肝素敏感试验(例如,已知pt试验对凝固抑制剂诸如肝素中等敏感)的实施方案中,可以将聚合物选择为中和肝素的聚合物诸如交联的或非交联的sbq-pva。在某些实施方案中,所述pva可以是光活化的stilbizonium盐。图10和11如下例证了该概念。将凝固传感器与大(图10)或小(图11)sbq-pva固定化的凝固底物一起直接印制。随后,将pt凝固活化剂印制在固定化底物基质的上面。然后用适当的流体试验这些传感器以评估凝固时间。与来自图5和6的结果一样,印制更多固定化底物基质的一个益处是,增加的量的固定化底物基质导致pa的增加。并且,当将肝素掺入全血样品中时,凝固时间没有象对它们预期的那样延长(图10和11(应答曲线181)(肝素波峰)应当更类似于应答曲线182(异常长的对照流体)凝固时间,但是实际上的表现更类似于没有肝素的全血(应答曲线183))。这些结果反映了聚合物(诸如sbq-pva)的肝素中和作用。此外,应用更大量的固定化基质会导致更大的中和作用(例如,对比图10和11中全血和含有肝素的全血之间的偏差(bias))。图10的数据代表大基质印制并表明当将肝素加入至1iu/ml时存在14%延伸,而在图11中,当沉积更小印制量的中和基质时,存在55%延伸。在图10和11中,掺入肝素的血液的表现更类似于未改变的全血(与它对异常长的对照流体的类似相比),从而反映了存在干扰物/肝素中和作用。该数据表明,增加pva印制的面积会减少肝素对pt测定的干扰物作用。图12提供的进一步实验证据表明,交联的sbq-pva可以用于给固定化的底物pt测定赋予肝素中和性质或肝素不敏感性。具体地,图12显示了在全血样品(应答曲线184)和掺入了0.4(应答曲线184)或1.2iu/ml的(应答曲线185)的肝素的全血样品(应答曲线185)上进行的pt测定的结果,其中不使用肝素酶(在pt试验中常规地包括用于中和肝素的试剂),但是将递增量的sbq-pva加入相同大小的印制中。数据表明,增加sbq-pva层的浓度会导致在有肝素存在下从全血的凝固时间延伸的下降。这表明,可以调节pt微环境传感器应答以在不使用昂贵肝素酶的情况下减少肝素的干扰。不受理论的约束,由交联的或非交联的sbq-pva赋予的正电荷似乎可以给固定化底物pt测定赋予肝素中和性质或肝素不敏感性。更具体地,sbq侧基是阳离子,pva是阴离子,且肝素是阴离子,因而假定通过带正电荷的sbq实现的在电极的定位区域中的斥力会从固定化的传感器微环境排斥肝素,或带正电荷的sbq会与带负电荷的肝素相互作用并废除肝素的作用于凝固因子上的能力。该理论被以下事实进一步证实:阴离子聚合物(诸如羟丙基纤维素)可以用于监测肝素疗法的诊断性凝固时间试验(例如,aptt和act),不会给测定赋予肝素中和性质或肝素不敏感性。在额外或替代实施方案中,所述聚合物可以是不中和肝素的聚合物,其然后可以随后经过处理或修饰以变成中和肝素的。例如,在其中要使用固定化的聚合物层进行的诊断性凝血试验具有肝素敏感性的实施方案中,可以将聚合物选择成包括至少一种不中和肝素的组分,例如,所述组分选自羟丙基纤维素和elvace、羧甲基纤维素、聚乳酸、聚乳酸、聚乳酸-乙醇酸共聚物、纤维素、纤维素衍生物、羟丙基甲基醋酸纤维素琥珀酸酯、菊糖、果糖、果聚糖、果聚糖衍生物和聚乙醇酸。所述不中和肝素的聚合物的一种或多种组分然后可以经过处理或修饰以产生中和肝素的层。在某些实施方案中,所述处理或修饰可以包括改变不中和肝素的聚合物的一种或多种组分的电荷,将肝素酶加入聚合物基质中,和/或将聚合物层构造成优先结合肝素上的硫酸酯基团。在额外或替代实施方案中,所述聚合物可以由不中和肝素的聚合物形成。例如,在其中要使用固定化的聚合物层进行的诊断性凝血试验用于监测肝素疗法(例如,aptt和act试验)的实施方案中,所述聚合物层可以包括至少一种不中和肝素的组分,所述组分任选地选自:羟丙基纤维素、elvace、羧甲基纤维素、聚乳酸、聚乳酸、聚乳酸-乙醇酸共聚物、纤维素、纤维素衍生物、羟丙基甲基醋酸纤维素琥珀酸酯、菊糖、果聚糖、果聚糖衍生物和聚乙醇酸。在包含自旋涂布(spincoating)的实施方案中,可以将固定化的底物和/或试剂聚合物层使用紫外线通过光刻法模印以使用掩模(mask)交联物质,随后除去未交联的物质,使得固定化的底物和/或试剂聚合物层被选择性地包被。在包含微分配(参见,例如,美国专利号5,554,339,其通过引用整体并入本文)的实施方案中,可以将适当量的每种涂层应用于区域,所述区域任选地被构造为围堵(containment)边界的另外结构组分限制。可替换地,表面处理(例如,向气体等离子体(plasmas)的暴露)可以用于控制表面能,且因而控制微分配的物质的铺展。在某些实施方案中,可以将一种或多种试剂和底物187固定化在聚合物层175内,如在图13a中所示。根据本发明的这些方面,包含一种或多种底物、聚合物(诸如可光变形的聚合物(例如,pva))和一种或多种试剂的水性底物-聚合物-试剂基质可以用于将所述一种或多种底物和所述一种或多种试剂固定化在转换器180上或附近。固定化的聚合物层175可以通过自旋涂布或通过微分配水性底物-聚合物-试剂基质来形成。在优选的实施方案中,用于aptt或act试验的一种或多种试剂或底物187可以固定化在聚合物层175内,且包含一种或多种试剂或底物187的固定化试剂-底物-聚合物层175的干燥体积可以是在约0.55-2.0nl的范围内,优选地在约1.0-1.5nl的范围内。在某些实施方案中,固定化的聚合物层175是基本上平面的且具有在约0.1-100μm的范围内的厚度。在额外或替代实施方案中,固定化的聚合物层175是基本上圆顶的且具有在约0.1-100μm的范围内的最大圆顶厚度。尽管在图13a中显示的试剂不同地定位在聚合物层175的中央区域中,在优选的实施方案中,所述试剂均匀地分散在底物-聚合物层中。在某些实施方案中,一种或多种试剂或底物187可以作为单独层形成在固定化的聚合物层175上面和/或附近,如在图13b和13c中所示。进一步,一种或多种试剂或底物可以一起或在分开的位置定位/固定化。根据本发明的这些方面,可以将一种或多种试剂或底物187旋转涂布或印制在固定化的聚合物层175(例如,pva层)上面和/或附近以将电化学或光信号定位在转换器180上面和/或附近。在优选的实施方案中,用于pt试验的一种或多种试剂或底物187可以与固定化的聚合物层175分开形成,且固定化的聚合物层175的干燥体积可以是在1.5-2.2nl的范围内,优选地在1.60-2.00nl的范围内。在某些实施方案中,固定化的聚合物层175是基本上平面的且具有在约0.1-100μm的范围内的厚度。在额外或替代实施方案中,固定化的聚合物层175是基本上圆顶的且具有在约0.1-100μm的范围内的最大圆顶厚度。传感器和芯片设计微制造的传感器阵列的一个优选实施方案包含至少一个转换器(例如,工作电极或光学检测器)。例如,所述微制造的传感器阵列可以包含一对微环境传感器或转换器,其包含第一微环境传感器或转换器(例如,pt传感器)和任选的第二微环境传感器或转换器(例如,aptt传感器)。在某些实施方案中,所述微环境传感器或转换器可以制造为分别在硅芯片上的靠近结构。在额外或替代实施方案中,除了第一微环境传感器或转换器和任选的第二微环境传感器或转换器以外,所述微制造的传感器阵列还可以包含一个或多个血液化学传感器。例如,所述传感器阵列还可以包含一个或多个被构造成测量钠、钾、钙、氯化物、二氧化碳、葡萄糖、血液尿素氮(bun)、肌酸酐、ph、co2分压、o2分压、乳酸盐、镁或另一种分析物中的一种或多种的传感器。在某些实施方案中,所述转换器可以形成为电极,其具有被光限定的聚酰亚胺层包被的金表面。例如,如在图14中所示可以实现传感器阵列的一个优选实施方案的晶片-水平微制造。平面的不导电底物190可以用作传感器阵列的基底。通过常规方式,例如,导电印制或本领域技术人员已知的用于形成至少一种晶体管的微制造技术,可以将导电层195沉积在底物190上。导电层195可以包含贵金属诸如金、铂、银、钯、铱或其合金,尽管也可以使用其它惰性金属(诸如钛和钨或其合金),还可以使用石墨、导电聚合物或其它物质的许多非金属电极。例如,基极可以包含在15μm中心上的5-10μm金薄片(例如,7μm金薄片)的正方形阵列。所述阵列可以覆盖具有大约300-900μm直径(任选地400-800μm或约600μm直径)的区域(例如,圆形区域),且可以通过光图样形成法在底物上面形成至多1.5μm厚度的聚酰亚胺或光致抗蚀剂的薄层,所述底物由一系列包含si、sio2、tiw和/或au或它们的组合的层制成。在某些实施方案中,所述基极具有约130,000-300,000平方μm的工作面积,直接在传感器上面的样品的体积可以是约0.1-0.3μl,且在芯片上面的样品的体积可以是1-3μl。根据本发明的这些方面,在基极的区域中的导管具有小于约6μl/约1平方mm、优选地小于约50mm/约2平方mm、更优选地小于约100μm/约500平方μm的体积与传感器面积比。因此,所述微电极阵列会提供可检测部分的高收集效率,所述可检测部分是具有减少的来自任何电化学背景电流(其与暴露的金属的电容有关)的贡献的电活性物种(electroactivespecies)。具体地,在绝缘聚酰亚胺或光致抗蚀剂层中的开口会限定金电极的区域,在此处电活性物种(例如,对-氨基苯酚)可以被氧化,诸如在每个分子两个电子的反应中。微制造技术(例如光刻技术和等离子体沉积)可以用于在有限空间中构建多层传感器结构。例如,用于在硅底物上微制造电化学免疫传感器的方法公开在美国专利号5,200,051(其特此通过引用整体并入)中,且包括,例如,分配方法,用于将底物和试剂附着于表面(包括光形成的层)的方法和用于进行电化学测定的方法。微制造的传感器阵列还可以包含电连接195和固定化的聚合物层205(如上面关于图4、7a、7b和7c讨论的),其沉积在导电层195和/或不导电底物190的至少一部分上。在本发明中,固定化的聚合物层205可以是包含具有可检测部分的、凝血酶可切割的肽的多孔聚合物层,所述肽被构造成通过产生能够被测量的变化而对活性凝血酶的存在做出应答。如在图15和16中所示,在某些实施方案中,所述微制造的传感器阵列可以包含硅芯片210,其包括位于硅芯片210的不同垂直面(a)和(b)上的微环境电流测定传感器或转换器215和220。传感器215可以通过线路225连接至第一电流测定针230(例如,暂时电连接器),且传感器220可以通过线路235连接至第二电流测定针240(例如,暂时电连接器)。在某些实施方案中,所述传感器215可以被构造为aptt传感器,且所述传感器220可以被构造为pt传感器,它们二者都形成在单个硅芯片210上且定位在现场护理试验筒的一个或多个导管内。如在图15中所示,传感器215可以被构造成具有靶十字线设计,所述靶十字线设计优选地包含在硅芯片210的上部区域中的多个同心环(例如,2、3、4或更多个同心环),且传感器220可以被构造成具有靶十字线设计,所述靶十字线设计优选地包含在硅芯片210的下部区域中的多个同心环(例如,2、3、4或更多个同心环)。具体地,基于传感器215和220中的每一个的印制和性能特征,选择在芯片210上的传感器215和220的设计和布置。但是,本领域普通技术人员应当理解,在不脱离本发明的精神和范围的前提下预见到传感器的任何设计或布置。此外,尽管在图15的实施例中的传感器215和220是电流测定传感器,但是可以使用其它电化学过程或光学过程,所述过程使用其它电化学或光学传感器。例如,光波波导器和电荷耦合器件(ccd)照相机芯片。例如,电位测定传感器可以用于检测离子物质诸如na+或k+。如本文中所述,电流测定传感器或转换器215和220可以形成为电极,所述电极具有暴露于导管的内部环境的金表面(例如,没有聚酰亚胺或光致抗蚀剂覆盖物)且被构造成直接接触放在导管内的生物样品。可以形成具有金表面的线路225和235,所述金表面被光限定的聚酰亚胺或光致抗蚀剂层包被,使得线路225和235绝缘而不暴露于放在导管内的生物样品。可以形成包含围堵环结构245和250的线路225和235,所述围堵环结构245和250被构造成含有固定化的试剂-底物-聚合物层。例如,固定化的试剂-底物-聚合物层(如上面关于图4、7a、7b和7c讨论的)可以沉积在位于围堵环结构245和/或250内的传感器215和/或220的至少一部分上。线路225和235分别终止于第一电流测定针230和第二电流测定针240,它们用于与分析仪或筒读数器(例如,如在美国专利号4,954,087中所述的筒读数器,其整个内容通过引用并入本文)中的连接器发生接触。在本发明的优选实施方案中,所述分析仪通过第一电流测定针230和第二电流测定针240在电流测定传感器215和220中的每一个与参比电极之间施加电势(在下面关于图17详细描述),并测量作为电化学信号由被切割的底物产生的电流变化。所述电化学信号与生物样品中产物的浓度成比例。电流测定传感器215和220具有相对于参比电极的大约+0.4v施加电势,且在另一个优选的实施方案中,电流测定传感器215和220具有相对于参比电极的大约+0.1v施加电势。在大约+0.1v由酶反应产物产生的信号可与在大约+0.4v由未反应的底物产生的信号辨别开。在使用与n-苯基-对苯二胺或n-[对甲氧基苯基-]-对苯二胺可检测部分连接的凝血酶可切割的肽tos-gly-pro-arg-、h-d-phe-pip-arg或bz-phe-val-arg的本发明实施方案中,在大约+0.4v的电压检测到完整底物。在大约+0.1v的电压检测到产电的反应产物n-苯基-对苯二胺或n-[对甲氧基苯基-]-对苯二胺。因而,在这些实施方案中,分析仪向电流测定传感器215和220施加电势,并产生与生物样品中的底物浓度成比例的电化学信号。并且,分析仪向电流测定传感器215和220施加电势,并产生与生物样品中的产物浓度成比例的电化学信号。在凝血酶将底物水解以后,形成在电流测定传感器215和220处反应的产物,并产生可与底物产生的信号辨别开的信号。应当指出,用于通过电流测定法检测底物和产物的确切电压将随底物和产物的化学结构而变化。重要的是,用于检测底物和产物的电压的差异是足够大的以防止读数之间的干扰。对于有些底物,电化学上检测底物所需的电压高得以致于超过在水性缓冲溶液中的实用测量。在这些情况下,仅要求所述产物可通过电流测定法检测。在某些实施方案中,在图15中所示的硅芯片210还可以包括多导管电导测定传感器255和260(例如,血细胞比容传感器)。电导测定传感器255和260被构造成确定生物样品到达和/或在电流测定传感器215和220处离开。更具体地,电导测定传感器255和260与导管或传感器导管的长度垂直定位,且每个传感器的电极对之间的电阻(electricalresistance)可以用于监测生物样品的流体前线(fluidfront)的相对位置。在末端处,开路读出指示生物样品已经被推出电流测定传感器215和220,且闭路读出指示电流测定传感器215和220被生物样品覆盖。如在图15中所示,电导测定传感器255可以包含至少2个定位在电流测定传感器215的中点的上游的电极265和270(即,第一电极对)。电极265和270可以通过线路275和280分别连接至电导测定低针285和ac源或电导测定高针290(例如,暂时电连接器)。可以形成具有金表面的线路275和280,所述金表面被光限定的聚酰亚胺或光致抗蚀剂层包被,使得线路275和280绝缘而不暴露于放在导管内的生物样品。电导测定传感器260可以包含至少2个定位在电流测定传感器220的中点的下游的电极295和300(即,第二电极对)。电极295和300可以通过线路275和280分别连接至电导测定低针285和ac源或电导测定高针290(例如,暂时电连接器)。这样,在某些实施方案中,所述流体在第一流体导管中到达第一电极对(例如,在到达电流测定传感器215之前),然后随后在第二流体导管中到达第二电极对(例如,在到达电流测定传感器220之后)。如在图16中所示,在另一个实施方案中,所述硅芯片210还可以包括包含至少2个电极302和303的第三电导测定传感器301。电极302和303可以通过线路304连接至第二ac源或电导测定高针305(例如,暂时电连接器)。根据本发明的这些方面,第三传感器的应用允许2个二元流体检测事件,例如,二者是关/开(off/on),这是可用电流电路和软件限制容易检测的。在2个电导率传感器的情况下(显示在图15中),电流电路和软件依赖于快速地连续检测样品的电阻的2个‘下降’的能力。通常,第一个下降是大的,因为它从干状态进入湿状态且电路是完整的。当样品在第二流体导管中到达时,电阻的第二下降小得多,且因此更难以与信号噪音和信号的小变化区分开。另外,每个电阻变化的振幅随样品性能而变化。因此和有利地,在某些实施方案中,具有3个电导测定传感器的布置允许使用电导测定传感器255(显示在图15中)和电导测定传感器301(显示在图16中)的2个可转换的电导率路径。如在图17中所示,在某些实施方案中,所述微制造的传感器阵列还可以包含包括参比传感器或电极307的接地芯片306。根据本发明的方面,其中传感器215和220是电流测定传感器,参比电极307可以被构造为对电极以完成电路。在一个优选的实施方案中,参比电极307可以包含沉积在固体底物(即,ag/agcl参比电极)上的银金属(ag)和它的银盐(agcl)。参比电极307可以通过线路308连接至参比针309(例如,暂时电连接器)。可以设计微制造的传感器阵列,使得接地芯片306定位在半导体芯片210的上游,如关于图15和16更详细地讨论的。但是,应当理解,传感器和接地芯片的其它布置是可能的,而不脱离本发明的精神和范围。例如,所述传感器阵列还可以包含一个或多个另外的传感器芯片(未显示),所述芯片被构造成检测可能感兴趣的各种分析物,诸如肌钙蛋白i、肌钙蛋白t、ckmb、原降钙素、bhcg、hcg、ntprobnp、probnp、bnp、肌红蛋白、甲状旁腺激素、d-二聚体、ngal、半乳糖凝集素-3和/或psa以及其它分析物。如在图18中所示,在优选的实施方案中,所述微制造的传感器阵列可以包含硅芯片310,其包括位于所述硅芯片310的相同垂直面(a)上的微环境电流测定传感器或转换器315和320。传感器315可以通过线路325连接至第一电流测定针330(例如,暂时电连接器),且传感器320可以通过线路335连接至第二电流测定针340(例如,暂时电连接器)。在某些实施方案中,所述传感器315可以被构造为aptt传感器,且所述传感器320可以被构造为pt传感器,它们二者都形成在单个芯片310上且定位在现场护理试验筒的导管内。如在图18中所示,可以构造具有圆环形设计的传感器315,其在用包含多个同心环(例如,2、3、4或更多个同心环)的靶十字线设计构造的传感器320的位置的上游位置。具体地,基于传感器315和320中的每一个的印制和性能特征,选择在芯片310上的传感器315和320的设计和布置。但是,本领域普通技术人员应当理解,在不脱离本发明的精神和范围的前提下预见到传感器的任何设计或布置。此外,尽管在图18的实施例中的传感器315和320是电流测定传感器,但是可以使用其它电化学过程或光学过程,所述过程使用其它电化学或光学传感器,例如,电位测定传感器可以用于检测离子物质诸如na+或k+。如本文中所述,传感器或转换器315和320可以形成为电极,所述电极具有暴露于导管的内部环境的金表面(例如,没有聚酰亚胺或光致抗蚀剂覆盖物)且被构造成直接接触放在导管内的生物样品。可以形成具有金表面的线路325和335,所述金表面被光限定的聚酰亚胺层包被,使得线路325和335绝缘而不暴露于放在导管内的生物样品。可以形成包含围堵环结构345和350的线路325和335,所述围堵环结构345和350被构造成含有固定化的试剂-底物-聚合物层。例如,所述固定化的试剂-底物-聚合物层(如上面关于图4、7a、7b和7c讨论的)可以沉积在位于围堵环结构345和/或350内的传感器315和/或320的至少一部分上。线路325和335分别终止于第一电流测定针330和第二电流测定针340处,所述电流测定针用于与分析仪或筒读数器(例如,如在美国专利号4,954,087中描述的筒读数器)中的连接器发生接触。在某些实施方案中,硅芯片310还包括集成的参比电极355。根据本发明的方面,其中传感器315和320是电流测定传感器,参比电极355被构造为对电极以完成电路。参比电极355可以包含沉积在固体底物上的银金属(ag)和它的银盐(agcl)(即,ag/agcl参比电极)。所述参比电极可以通过线路360连接至ac地线和参比针365(例如,暂时电连接器)。可以形成具有金表面的线路360,所述金表面被光限定的聚酰亚胺或光致抗蚀剂层包被,使得线路360绝缘而不暴露于放在导管内的生物样品。在优选的实施方案中,如在图18中所示以西洋跳棋盘(checkboard)图案设计参比电极355以提高参比电极355的表面的润湿性。具体地,已经意外地发现,使用西洋跳棋盘图案可以提高参比电极355的润湿性,因为agcl是相对疏水的且可以在使用agcl的固体贴片时促进气泡在参比电极355的表面上的形成,这会导致差电路。如上面关于硅芯片310详细讨论的和如在图19中所示,在本发明的优选实施方案中,所述分析仪通过第一电流测定针330和第二电流测定针340在电流测定传感器315和320与参比电极355之间施加电势,并测量作为电化学信号由被切割的底物产生的电流变化。所述电化学信号与生物样品中产物的浓度成比例。电流测定传感器315和320具有相对于参比电极355的大约+0.4v施加电势,且在另一个优选的实施方案中,电流测定传感器315和320具有相对于参比电极355的大约+0.1v施加电势。在大约+0.1v由酶反应产物产生的信号可与在大约+0.4v由未反应的底物产生的信号辨别开。回去参考图18,在某些实施方案中,硅芯片310还可以包括电导测定传感器370和375(其还可以作为血细胞比容传感器起作用)。电导测定传感器370和375可以分离以形成2个传感器对,在芯片310的每个末端各一个。电导测定传感器370和375分别被构造成确定生物样品到达和/或在电流测定传感器315和320处离开。更具体地,电导测定传感器370和375位于与导管或传感器导管的长度垂直的弧中,且每个传感器的电极对之间的电阻可以用于监测生物样品的流体前线的相对位置。在末端处,开路读出指示生物样品已经被推出电流测定传感器315和320,且闭路读出指示电流测定传感器315和320被生物样品覆盖。如在图20中所示,电导测定传感器370可以包含至少2个相对彼此在预定距离(d1)处定位的电极380和385(即,第一电极对)。在某些实施方案中,电导测定传感器370可以相对于电流测定传感器315的中点(v)定位在硅芯片310上(例如,相对于中点(v)在上游、下游或线内)。电极380可以通过线路390连接至ac源针395(例如,暂时电连接器)。电极385可以通过线路400、参比电极355和线路360连接至ac地线和参比针365。可以形成具有金表面的线路390和400,所述金表面被光限定的聚酰亚胺或光致抗蚀剂层包被,使得线路390和400绝缘而不暴露于放在导管内的生物样品。电导测定传感器375可以包含至少2个相对彼此在预定距离(d2)处定位的电极405和410(即,第二电极对)。在某些实施方案中,电导测定传感器375可以相对于电流测定传感器320的中点(x)定位在硅芯片310上(例如,相对于中点(x)在上游、下游或线内)。电极405可以通过线路415、参比电极355和线路360连接至ac地线和参比针365。电极410可以通过线路420和线路390连接至ac源针395。可以形成具有金表面的线路415和420,所述金表面被光限定的聚酰亚胺或光致抗蚀剂层包被,使得线路415和420绝缘而不暴露于放在导管内的生物样品。在优选的实施方案中,电导测定传感器370和375分别被构造成检测导管内的生物样品在电流测定传感器315和320处的到达。如在图21中所示,基于第一电阻下降425(当生物样品到达电导率传感器370时)和第二电阻下降430(当生物样品到达电导率传感器375时)的确定,可以检测生物样品在电流测定传感器315和320处的到达。在额外或替代实施方案中,在电导测定传感器370和375中的任一个或两个处的电阻的升高或波峰(未显示)的确定,可以用于检测气泡在导管内的存在,所述导管位于电流测定传感器315和320中的任一个或两个上。电导测定传感器370和375的电阻曲线应当优选地提供2个确定定义的大致相等振幅的电阻下降。在某些芯片设计中,如在图22a中所示,电导测定传感器435和440可以被构造为在各个电流测定传感器445和450附近的芯片的相对末端上的单独条。但是,发现这样的设计的电阻曲线455经常包括额外阶梯460,其可归因于由于参比电极465的疏水性而在参比电极465上暂时停止的样品。应当理解,这可能使得难以将第二电阻下降译解为参比电极465或到达第二电导测定传感器440的样品的润湿。另外,两个阶梯之间的时间是十分短的,从而使得难以计时,且第二到达的电阻下降与第一下降相比小得多,从而使得难以检测。因此,如在图22b中所示,在本发明的优选实施方案中实现的芯片设计利用电导测定传感器370和375,它们各自拆分成分别包含至少2个间隔预定距离(d1)和(d2)的电极380、385和405、410。如在电阻曲线470中所示,主要电阻下降425和430发生在两对电导测定传感器370和375处。因而,减少从参比电极355的润湿观察到的额外的电阻下降460(显示在图22a中)的影响。进一步,将电导测定传感器370和375定位在芯片的前部和后部处来增加电阻下降425和430之间的时间以更好地区分电阻下降425和430。此外,在某些实施方案中,在电极380和385之间提供的间隔或预定距离(d1)是大于在电极405和410之间提供的间隔或预定距离(d2)的值“n”,使得第二电阻下降430的振幅与替代芯片设计的电阻下降475相比增加(显示在图22a中)。例如,可以将(d1)构建为(d2)2倍以实现第二电阻下降的振幅的约1000欧姆增加。与图22a中所示的芯片设计的电阻下降的比率相比,(d1)相对于(d2)的增加会有效地增加图22b中所示的芯片设计的电阻下降的比率。有利地,电阻下降的这种增加允许在开启/向前(on/forward)马达或泵位置480过程中更好地检测生物样品在电导测定传感器370和375处的到达。在某些实施方案中,本发明的方法可以包括在芯片上以受控的速度向前和向后连续移动生物样品。控制电导测定传感器370和375保持为开路和闭路的时间会控制生物样品改变方向时的位置。例如,在分析仪内的气动泵可以构造成振荡导管中的生物样品,其中生物样品的后缘定位在电导测定传感器370的区域中,以便溶解靠近所述后缘的样品的该部分中的底物。所述振荡可以是在0.2-10赫兹的范围内的频率,持续1-100秒的范围内的时间段。在一种优选的方法中,所述振荡可以是在约1.5赫兹的范围内的频率,持续约20秒的时间段。在另一种优选的方法中,所述振荡可以是在约0.3赫兹的频率,且电流测定传感器315和320(如在图20中所示)可以构造成在每个振荡处产生信号。如果红细胞存在于生物样品中,所述振荡可以是在足以防止红细胞沉降在电流测定传感器315和320上的频率。在某些实施方案中,每当生物样品被振荡经过电流测定传感器315和320时电流测定传感器315和320确定产物的浓度。例如,对于电流测定传感器315和320中的每一个,第一电流测定传感器信号可以由分析仪存储,且来自电流测定传感器315和320的随后信号可以被存储并与第一个和其它存储的信号进行对比以便确定电流测定传感器信号的最大变化速率。然后可以分析这些数据点以确定电流测定传感器信号的最大变化速率的固定分数。这些数据点因而可以用于确定电流测定传感器315和320中的每一个的目标凝固参数。在替代实施方案中,传感器或转换器可以形成为光学检测器,例如ccd照相机芯片和光波波导器。所述光学检测器可以是来自可检测部分的荧光、化学发光或生物发光发射的检测器或可检测部分的吸光度的检测器。在这样的实施方案中,所述可检测部分可以是光学染料、荧光发射体、化学发光发射体或生物发光发射体。在其它实施方案中,所述传感器或转换器可以形成为试验条,例如,葡萄糖试验条,如在美国专利申请号13/724,348(其以它的整体并入本文)中所述。例如,可以在本文描述的筒内包括试验条。在某些实施方案中,可以将样品手工地放在试验条上,且这样,不需要与这样的实施方案一起包括本文描述的微流体系统。如本领域众所周知的,葡萄糖试验条装置可以包括被动毛细管流体元件以将样品递送至传感器或传感器阵列。这样,葡萄糖试验条的元件、部件和功能性可以适合本发明,而不脱离本发明的精神和范围。用于样品分析的系统和方法如在图23中所示,本发明的系统500可以包含自含式的一次性传感装置或筒505和读出装置或仪器510(例如,分析仪)。在某些实施方案中,筒505是一次性使用的装置,其被构造成在一次性使用以后抛弃。将要测量的流体样品(例如,全血)抽入筒505的样品进入孔口或端口515中,可以将筒505穿过带槽的开口520插入读数器510中。读数器510可以包含处理器,其被构造成进行分析物浓度的测量、电阻的测量、鉴别芯片要测量的分析物或分析物集合、和/或流体样品内的诊断性凝固时间的确定,如本文中更详细地讨论的。经由读数器510上的端口535至计算机端口540,可以将由读数器510进行的测量和确定输出至显示器525或其它输出装置,诸如打印机或数据管理系统530。传输可以是通过wifi、蓝牙连接、红外等。在其中筒505(例如,微环境传感器)内的传感器545是基于电化学运行原理的实施方案(例如,第一传感器和任选的第二传感器)中,可以构造成通过电连接器550与读数器510发生电接触。例如,所述连接器可以具有在共同拥有的美国专利号4,954,087(通过引用整体并入本文)中公开的设计。在某些实施方案中,所述pt和aptt传感器可以被构造成通过电连接器550与读数器510内的试验表的电连接器连接(参见,例如,美国专利号5,096,669和4,954,087,通过引用整体并入本文)。读数器510还可以包括筒505中的自动流体流动补偿的方法,如在共同拥有的美国专利号5,821,399(其也通过引用整体并入本文)中公开的。在一个实施方案中,如在图24中所示,自含式的一次性传感装置或筒555可以包含盖子560、基底565和安置在基底565和盖子560之间的薄膜粘着垫圈(未显示)。筒555可以被构造成用于插入读数器510中,且因此筒555可以包含多个用于此目的的机械和电连接(未显示)。有利地,筒555的一个特征是,一旦流体或生物样品被加载进筒555内,流体或生物样品的分析可以结束,且可以在操作员或其他人不接触流体或生物样品的情况下抛弃筒555。参考图24,盖子560可以由刚性材料(优选塑料)制成,且能够在不破裂的情况下在柔性铰链区570、575和580处重复变形。盖子560可以包含盖585,其通过柔性铰链570连接至盖子560的主体。在运行中,通过样品进入端口595将流体或生物样品引入样品容纳室590中以后,可以将盖585在入口上面固定至样品进入端口595,从而防止样品渗漏。盖585可以由钩600保持就位。盖子560还可以包含2个可变形部件605和610,所述部件605和610可相对于盖子560的主体移动且其可以通过柔性铰链区575和580连接至盖子560。可变形部件610可以被构造成由第一泵送装置运行,从而对由腔体615和垫圈构成的气囊施加力。可变形部件610的运行会转移筒555的导管内的流体。可变形部件605可以被构造成由第二泵送装置运行,从而对垫圈施加力,所述垫圈可以因为在其中切出的裂缝而变形。在某些实施方案中,垫圈的变形可以将压力传送到位于腔体620中的装满流体(例如,大约130μl分析/洗涤溶液、对照流体或校正流体)的含流体的箔包上,使所述箔包破裂,并将流体驱逐进导管625中用于随后在样品分析过程中用在其它导管中。应当理解,尽管凝固测定形式通常不要求使用这些流体,但是在将凝固试验与其它试验组合的单个装置中通常可能需要流体,例如,在分析物(诸如bnp和肌钙蛋白)的免疫测定中的洗涤流体,和在化学试验(诸如钾、肌酸酐和葡萄糖)中的校正流体。在替代实施方案中,垫圈的变形可以将压力传送到由腔体620构成的气囊上用于移动筒555的导管内的流体。在其它实施方案中,第二泵送装置可能不作用于腔体620,作为替代,腔体620可以被构造为废物室。由在读数器510(关于图23讨论)内施加于筒555的机构产生的筒555中的额外作用可以用于将一个或多个空气段在样品容纳室590和导管630内的受控位置处注射进流体或生物样品中。如免疫测定操作领域的普通技术人员会理解的,所述空气段可以用于用最小量的流体洗涤传感器阵列的传感器表面和周围导管630(例如,有限的洗涤循环,其中洗涤体积可以小于流体或生物样品的体积的50倍,和/或少于3个独立循环的清洁洗涤缓冲液(例如,3个使用新鲜洗涤缓冲液的独立洗涤步骤))。例如,盖子560还可以包含被柔软薄膜覆盖的孔。在运行中,施加于所述膜的压力可以将一个或多个空气段通过垫圈中的小孔驱逐进导管630中。在某些实施方案中,导管630的横截面面积可以是在约0.1mm2至约10mm2的范围内。在某些实施方案中,盖子560的下表面还包含样品容纳室590、导管630和另一个导管635(例如,废物导管)。样品容纳室590和导管630可以包括一个或多个阻塞物(constriction)或毛细管档栓(stop)640和642,其通过向流体或生物样品的流提供阻力来控制流体流。任选的涂层(未显示),例如,干燥试剂涂层,可以在导管630上提供疏水表面,其与垫圈孔一起控制样品容纳室590和导管635之间的流体流。样品容纳室590可以被构造成将样品进入端口595连接至组装的筒555中的导管630。根据其中存在多个芯片(例如,接地芯片和传感器芯片)的本发明的方面,切掉部分(cutaway)645可以容纳一个或多个传感器芯片650,所述传感器芯片650包含至少一个传感器655(例如,pt、aptt或act微环境传感器)或应答表面、以及任选的一个或多个电导测定传感器660。如果需要的话,切掉部分665可以容纳包含接地电极675的接地芯片670作为电化学传感器的返回电流路径,且也可以容纳任选的电导测定传感器。根据其中仅存在单个芯片(例如,组合的接地和传感器芯片)的本发明的方面,不可以与筒555一起包括切掉部分665和接地芯片670。在某些实施方案中,可以提供包含样品容纳室590的计量装置,所述样品容纳室590以阻塞物或毛细管档栓640为边界且沿着样品容纳室590长度具有来自包含包含腔体615的囊的空气进入点680。在进入点680处施加的空气压力驱动计量体积的样品经过阻塞物或毛细管档栓640。因此,由空气进入点680和阻塞物或毛细管档栓640之间的样品容纳室590的体积可以预先确定样品的计量体积。当可变形部件605被置换时,可以将与该体积对应的量的样品置换进导管630中。因此,这种布置可以提供计量装置用于将计量的量的未计量样品递送进筒555的各个下游导管中。在某些实施方案中,如果需要量化分析物,所述计量可以是有利的。因而,操作员可以免于在测量之前准确地测量样品的体积,从而节省时间、劳作并增加准确度和再现性。在优选的实施方案中,本发明是一种使用筒来确定全血样品中的诊断性凝固时间的方法。所述方法可以包括将未计量的流体样品穿过样品进入端口595引入筒555的样品容纳室590中(如在图24中所示)。毛细管档栓640阻止流体样品在该阶段进入导管630中,且样品容纳室590被样品填充。封闭盖585以防止流体样品从筒555渗漏。然后可以将筒555插入读出装置或设备510中,如在图23中所示且进一步公开在美国专利号5,821,399中,其通过引用整体并入本文。在某些实施方案中,筒向读出设备510中的插入会活化机构,其刺穿位于腔体620中的含有流体的包(当所述包被压靠在刺突(未显示)上时)。由此可以将流体驱逐进一个或多个导管(例如,导管630)中依次到达传感器区域。此后,泵装置(例如,气动泵)的运行将压力施加于由腔体615构成的气囊,从而迫使空气穿过导管在空气进入点680处进入样品容纳室590。毛细管档栓640限定原始流体样品的计量部分的边界。在由腔体615构成的气囊内产生的空气压力然后通过毛细管档栓640驱逐样品的计量部分。所述样品进入导管630中并与一种或多种试剂、一种或多种底物(例如,固定化的试剂-底物-聚合物层)和/或一个或多个传感器发生接触,所述传感器包含一个或多个转换器和任选的位于切掉部分665内的参比电极。也如在图24中所示,为了促进(i)一种或多种试剂在流体样品中的扩散,或样品在含有试剂的聚合物层中的扩散(取决于具体实施方案),(ii)两个途径之一对凝固级联的活化以产生凝血酶,(iii)活性凝血酶通过固定化的底物和/或试剂聚合物层的扩散,(iv)凝血酶可切割的肽的切割,(v)可检测部分的活化,和/或(vi)至少一个转换器对可检测部分的检测,可以将流体样品定位在导管630内以接触一种或多种试剂和/或底物、一个或多个固定化的聚合物层和/或一个或多个传感器(例如,微环境传感器)持续预定的时间阶段。筒的应用在本文中由具体实施方案来举例说明,其中确定流体样品(将其引入筒的样品容纳室中,随后将筒插入筒读出装置中)的诊断性凝固时间。所述筒读出装置通过垫子与电极/传感器发生电接触,并进行某些诊断试验。所述诊断试验使用电导率电极确定流体或样品是否存在于导管中;确定电短路是否存在于电极中;和确保传感器和接地电极在测定循环之前热平衡至优选的37℃。在优选的实施方案中,可以使用计量部分的流体样品(优选地在4-200μl之间,更优选地在4-20μl之间,且最优选地7μl)来进行测定,而其子体积(0.1-3.5μl之间)可以用于接触电极/传感器。将流体样品相对于传感器区域定位,使得流体样品的一部分位于一种或多种试剂、一种或多种底物(例如,固定化的聚合物层)和一个或多个传感器上,所述传感器包含一个或多个转换器和接地电极。在630的上或下段中(或在例如图24、25、26、31、34、36的测定导管中的任一个中)振荡预定的时间阶段(例如,0-10秒)以后,样品可以移动至第二导管或区域用于随后的混合或相互作用,或在信号产生之前变得静止或变得锁定在导管或筒内。在传感器芯片上的一个或多个电导率传感器可以用于控制关于图20、21、22a和22b讨论的这些过程。在随后的流体拆分或转移中,可能存在穿过控制压力或大小的元件的通道。这些方面在以后更详细地描述。在样品和传感器之间的接触时间中,(i)修正试剂有时间扩散进流体样品中或流体样品有时间扩散进修正试剂(在某些实施方案中,其可以被固定化)中,以便促进两个途径之一对凝固级联的活化以产生凝血酶,(ii)活性凝血酶有时间扩散穿过底物层(例如,固定化的底物和/或试剂聚合物层)并切割凝血酶可切割的肽,和(iii)活化的可检测部分有时间被至少一个转换器检测到。流体功能和筒的构型在优选的实施方案中,提供了一次性筒构型,其能够使2个物理上分离的试验在同一个一次性筒内在单个全血样品上同时或先后进行。一次性筒构型的元件除了包括来自分析仪的主动机构(例如,泵)以外还包括被动流体部件(例如,阀门、电阻和流体锁定元件)的使用以将样品分进单独的导管/区域中,使得每个样品段可以随后移动至特定传感器。本文中讨论了许多单独的构型,其允许将样品维持在单个通道中、将样品分入单独的流体导管、控制每个导管中的流体移动,例如,以将干燥的试剂和/或底物混合进样品段中,和/或随后将样品停留(和锁定)在传感器上用于分析。但是,应当理解,可以做出构型的各种修改、置换、省略和变化,而不脱离本发明的精神和范围。在下面的每个实施方案中,使用者可以将样品插入筒的入口室中。然后将筒关闭并插入分析仪中。使用作为筒中的气囊形成的隔膜泵和分析仪中的机械柱塞(如关于图24讨论的)来移动样品穿过筒。关于图25-29讨论的本发明的实施方案被构造成分割单个生物(例如,全血)样品并允许在两个导管(干燥的试剂和/或对每种试验特异性的底物位于其中)中的至少两段样品的独立混合控制。根据本发明的方面,所述底物可以是或不是定位(例如,根据一些实施方案,可以是或不是固定化)在传感器上面。溶解在样品中的试剂和/或底物保留在形成它们的导管内,因此消除试验之间的任何潜在交叉干扰。对于其中可能存在化学或物理干扰的任何两个试验(例如,凝固试验),这是多路技术的一个重要要素。如在图25中所示,本发明的一些实施方案涉及筒700的接地传感器第一构型,其中导管705在接地芯片720之前或上游在连接部707处分成第一导管710和第二导管715。第二导管715可以包含阻塞物或毛细管档栓725且被构造成经过包含至少一个分析物检测电极的传感器芯片730(如关于图15和16描述的)的下部区域。第一导管710被构造成经过接地芯片720(例如,如关于图17描述的具有参比传感器的接地芯片)和包含至少一个分析物检测电极的传感器芯片730(如关于图15和16描述的)的上部区域。筒700还可以包含定位在第一导管710内的至少一个流体锁定机构735(例如,膜海绵阀、微通道毛细管或微阵列阀)和一个或多个导管740(例如,排气道(vent)),所述导管740从第一导管710和第二导管715连通至腔体747。在该实施方案中,腔体747被构造为废物室(如关于图24讨论的)。但是,本领域技术人员应当理解,一个或多个导管740可以被构造成连通至废物导管(如关于图24讨论的)。图26显示了筒750的替代性接地传感器第一构型,其中导管755在接地芯片775之前或上游在连接部760处分成第一导管765和第二导管770。第二导管770可以包含阻塞物或毛细管档栓780且被构造成经过包含至少一个分析物检测电极的传感器芯片785(如关于图15和16描述的)的下部区域。第一导管765被构造成经过接地芯片775(例如,如关于图17描述的具有参比传感器的接地芯片)和包含至少一个分析物检测电极的传感器芯片785(如关于图15和16描述的)的上部区域。筒750还可以包含定位在第一导管765内的至少一个流体锁定机构790和一个或多个导管793和795(例如,排气道),所述导管793和795分别从第一导管765和第二导管770连通至废物导管797(如关于图24讨论的)。如在图27中所示,在筒700和750的运行中,使用双向隔膜泵(如关于图24描述的)将流体或生物样品从入口或样品容纳室移动至导管705/755。所述生物样品在连接部707/760(例如,t-连接部)处分成第一部分和第二部分。在优选的实施方案中,定位在第二导管715/770内的阻塞物或毛细管档栓725/780(例如,毛细管爆裂阀(burstvalve)或流体阻力(resistance)/阻塞物)造成样品优先填充第一导管710/765并在接地芯片720/775和传感器芯片730/785的上部区域内的至少一个电极(例如,aptt电极)上面移动。隔膜泵因此可以在第一导管710/765中来回移动样品的第一部分以将试剂和/或底物溶解并混合在样品中,而第二导管715/770中的样品的第二部分既不退出第二导管715/770也不移动至传感器芯片730/785。一旦已经在第一导管710/765中实现充分混合,样品的第一部分在传感器芯片730/785上面被推至流体锁定机构735/790(例如,在双面胶或模塑塑料部件(component)之一中形成的膜“海绵阀”或微通道),其提供压力阻力并将样品的第一部分有效地锁定在第一导管710/765中。此后可以开始第一导管710/765中的分析。随着第一导管710/765中的压力阻力显著地增加,剩余的样品的第二部分被迫穿过第二导管715/770中的阻塞物或毛细管档栓725/780。类似的来回混合过程然后可以应用于第二导管715/770。一旦将试剂和/或底物混合进样品的第二部分,可以将样品的第二部分定位于传感器芯片730/785上并可以开始第二导管715/770中的分析。例如,所述第二部分可以在传感器芯片730/785的下部区域内的至少一个电极(例如,pt电极)上面移动穿过第二导管715/770。根据本发明的方面,在有或没有使用关于图2、3、4、7a、7b、7c和9讨论的布置中的一种或多种固定化试剂/底物的情况下,可以形成在传感器芯片730/785的上部和下部区域内的电极。如在图28中所示,在筒700和750的另外运行过程中,使用双向隔膜可以使流体或生物样品从入口或样品容纳室移动至传感器芯片730/785(如关于图24描述的)。另外,一旦将试剂和/或底物混合进样品的第二部分中,样品的第二部分在传感器芯片730/785上面被推至另外的流体锁定机构745/798(显示在图25和26中)(例如,在双面胶或模塑塑料部件之一中形成的膜“海绵阀”或微通道),其提供压力阻力并将样品的第二部分有效地锁定在第二导管715/770中。一旦样品的第二部分被锁定在传感器芯片730/785上面的位置,可以开始在第二导管715/770中的分析。如在图29中所示,在筒700和750的另外运行过程中,使用双向隔膜可以使流体或生物样品从入口或样品容纳室移动至导管705/755。但是,在样品移动至连接部707/760之前,样品可以移动穿过另外的连接部(例如,t-连接部)(未显示在图25和26中)。另外的连接部被构造成将第一导管710/765和第二导管715/770与减压导管(reliefconduit)(例如,排气道)分离,所述减压导管具有阻塞物或毛细管档栓(例如,毛细管爆裂阀或流体阻力/阻塞物)以使穿过连接部707/760的样品流转向。样品的任何残余压力或移动然后前进到减压导管中。在优选的实施方案中,设计所述减压导管中的另外阻塞物或毛细管档栓,使得它具有比第一导管710/765和/或第二导管715/770中的流体锁定部件更低的压力阻力。如在图30中所示,通过使用多导管电导测定传感器确定和维持样品的定位(如本文中讨论的),在电极上面振荡样品的第一部分和第二部分,可以完成导管内的混合。在某些实施方案中,首先混合生物样品的第一部分以开始生物样品的第一部分与第一导管内的试剂和/或底物之间的反应,然后开始生物样品的第二部分与第二导管内的试剂和/或底物之间的反应。例如,aptt试验常规地要求比pt试验更长的试验时间,且因而在第一导管内进行的aptt试验可以比在第二导管内进行的pt试验更早地开始,使得所述试验大约同时完成。应当理解,筒700和750的接地传感器第一设计有利地提供了单个筒,其能够在两个单独导管内同时或先后进行两个独立测定(例如,pt和aptt)。在其中需要混合或混合有利的实施方案中,筒700和750的部件允许在第一导管和第二导管内的独立混合控制,无需担忧在一种或多种试剂已经变得暴露于生物样品以后级联途径的交叉活化或其它交叉电极干扰,因为所述传感器通过至少第一导管和第二导管的使用彼此物理上分离。关于图31-35讨论的本发明的实施方案被构造成将单个生物(例如,全血)样品分成两个导管(干燥的试剂和/或对每个试验特异性的底物位于其中)中的两段。关于图31-35讨论的构型与图25-29的构型之间的主要差异是,在图31-35的构型中,样品被在传感器上面推动并锁定就位,且位于传感器上的试剂和/或底物被特别设计成通过被动扩散溶解在样品中或保留在固定化层内。在进行凝固分析的实施例中,级联途径的活化及其检测发生在紧密靠近传感器的高浓度区域处。溶解在样品中或被包含在固定化的层中的试剂和/或底物保持在它们所印制的导管内,因此消除试验之间的任何潜在交叉干扰(数据未显示)。对于其中可能存在化学或物理干扰的任何两个试验(例如,凝固试验),这是多路技术的一个重要要素。如在图31中所示,本发明的一些实施方案涉及用于筒800的集成的接地传感器和分裂的传感器导管设计,其中导管805在接地/传感器芯片825之前或上游在连接部810处分成第一导管815和第二导管820。第一导管815被构造成经过接地/传感器芯片825(如关于图18讨论的)的第一区域,所述第一区域包含参比电极的一部分和至少一个分析物检测电极(例如,aptt电极)。第二导管820包含阻塞物或毛细管档栓822且被构造成经过传感器芯片825(如关于图18讨论的)的第二区域,所述第二区域包含参比电极的另一部分和至少一个其它分析物检测电极(例如,不同的分析物检测电极,诸如pt电极)。根据本发明的方面,在有或没有使用关于图2、3、4、7a、7b、7c和9讨论的布置中的一种或多种固定化试剂/底物的情况下,可以形成在第一导管815和第二导管820内的分析物检测电极。筒800还可以包含分别定位在第一导管815和第二导管820内的至少两个流体障碍机构830和835(例如,流体锁定机构、毛细管档栓或流体阻塞物)和一个或多个导管840和845(例如,排气道),所述导管840和845分别从第一导管815和第二导管820连通至腔体850。在该实施方案中,腔体850被构造为废物室(如关于图24讨论的)。但是,在替代实施方案中,一个或多个导管840和845可以被构造成连通至废物导管(如关于图24讨论的)。如在图32中所示,在筒800的运行中,使用双向隔膜泵(如关于图24描述的)使流体或生物样品从入口或样品容纳室移动至导管805。生物样品在连接部810(例如,t-连接部)处分成第一部分和第二部分。在优选的实施方案中,定位在第二导管820内的阻塞物或毛细管档栓822(例如,毛细管爆裂阀或流体阻力/阻塞物)造成样品优先填充第一导管815并在接地/传感器芯片825的第一区域上面移动。样品的第一部分被在接地/传感器芯片825上面推至流体障碍机构830(例如,在双面胶或模塑塑料部件之一中形成的膜“海绵阀”或微通道),其提供大于第二导管820的压力阻力,并将样品的第一部分有效地锁定在第一导管815中。此后可以开始第一导管815中的分析。随着第一导管815中的压力阻力显著地增加,剩余的样品第二部分被迫穿过第二导管820中的阻塞物或毛细管档栓822。类似地,样品的第二部分被在接地/传感器芯片825上面推至流体障碍机构835(例如,在双面胶或模塑塑料部件之一中形成的膜“海绵阀”或微通道),并将样品的第二部分有效地锁定进第二导管820中。此后可以开始在第二导管820中的分析。如在图33中所示,在筒800的另外运行过程中,使用双向隔膜可以使流体或生物样品从入口或样品容纳室移动至导管805。但是,在样品移动至连接部810之前,可以穿过另外的连接部855(例如,t-连接部)移动样品(显示在图31中)。另外的连接部855(例如,t-连接部)被构造成将第一导管810和第二导管820与减压导管860(例如,排气道)分离,所述减压导管860具有阻塞物或毛细管档栓(例如,毛细管爆裂阀或流体阻力/阻塞物)以使穿过连接部810的样品流转向。样品的任何残余压力或移动然后前进到减压导管860中。在优选的实施方案中,设计所述减压导管中的另外阻塞物或毛细管档栓,使得它具有比第一导管815和第二导管820中的流体障碍机构更低的压力阻力。本领域普通技术人员应当理解,用于筒800的集成的接地传感器和分裂的传感器导管设计有利地提供了单个筒,其能够在两个单独导管内同时或先后进行两个独立测定(应当理解,所述测定可以是不同的或相同的,例如,pt和aptt、pt和pt、aptt和aptt等),而不需要将生物样品与试剂和/或底物混合。根据该实施方案的方面,筒800的部件允许在第一导管815和第二导管820内进行两个单独分析试验,无需担忧在一种或多种试剂已经变得暴露于生物样品以后级联途径的交叉活化或其它交叉电极干扰,因为所述电极通过至少第一导管815和第二导管820的使用彼此物理上分离。此外,用于筒800的集成的接地传感器设计提供了比上面描述的接地传感器第一设计更简单的、更紧凑的筒设计,因为所述设计消除了完全分离接地传感器的空间要求和使生物样品移动至单独接地传感器所需的导管的额外长度。如在图34中所示,本发明的替代实施方案涉及筒870的集成的接地传感器和分裂的导管设计,其包含具有连接部880(例如,t-连接部)的导管875。连接部880被构造成将第一传感器导管885和第二导管890与减压导管895(例如,排气道)分离。第一传感器导管885被构造成经过接地/传感器芯片900(如关于图18讨论的)的第一区域,所述第一区域包含参比电极的一部分和至少一个分析物检测电极(例如,aptt电极)。第二传感器导管890被构造成经过传感器芯片900(如关于图18讨论的)的第二区域,所述第二区域包含参比电极的另一部分和至少一个不同的分析物检测电极(例如,pt电极)。根据本发明的方面,在有或没有使用关于图2、3、4、7a、7b、7c和9讨论的布置中的一种或多种固定化底物的情况下,可以形成在第一传感器导管885和第二传感器导管890内的分析物检测电极。筒870还可以包含分别定位在第一传感器导管885和第二传感器导管890内的至少两个流体障碍机构905和910和一个或多个导管915和920(例如,排气道),所述导管915和920分别从第一传感器导管885和第二导管890连通至腔体925。在该实施方案中,腔体925被构造为废物室(如关于图24讨论的)。但是,在替代实施方案中,一个或多个导管915和920可以被构造成连通至废物导管(如关于图24讨论的)。如在图35中所示,在筒870的运行中,使用双向隔膜泵(如关于图24描述的)使流体或生物样品从入口或样品容纳室移动至导管875并穿过连接部880。减压导管895(例如,排气道)具有阻塞物或毛细管档栓(例如,毛细管爆裂阀或流体阻力/阻塞物)以使样品流转向第一传感器导管885和第二传感器导管890。样品的第一部分被在接地/传感器芯片900上面推至流体障碍机构905(例如,在双面胶或模塑塑料部件之一中形成的膜“海绵阀”或微通道),所述流体障碍机构905将样品的第一部分有效地锁定在第一传感器导管885中。此后可以开始在第一导管885中的分析。样品的第二部分被在接地/传感器芯片900上面推至流体障碍机构910(例如,在双面胶或模塑塑料部件之一中形成的膜“海绵阀”或微通道),所述流体障碍机构910将样品的第二部分有效地锁定在第二传感器导管890中。此后可以开始在第二导管890中的分析。样品的任何残余压力或移动然后将前进到减压导管895中。在优选的实施方案中,设计在减压导管中的阻塞物或毛细管档栓,使得它具有比在第一传感器导管885和第二传感器导管890中的流体障碍机构更低的压力阻力。应当理解,用于筒870的替代性的集成的接地传感器和分裂的导管设计有利地提供了单个筒,其能够在两个单独传感器导管内同时或先后进行两个独立测定(应当理解所述测定可以是不同的或相同的,例如,pt和aptt、pt和pt、aptt和aptt等),而不需要将生物样品与试剂和/或底物混合。根据该实施方案的方面,筒870的部件允许在第一传感器导管885和第二传感器导管890内进行两个单独分析试验,无需担忧在一种或多种试剂已经变得暴露于生物样品以后级联途径的交叉活化或其它交叉电极干扰,因为所述电极通过至少第一传感器导管885和第二传感器导管890的使用彼此物理上分离。此外,筒870的集成的接地传感器设计提供了比上述的接地传感器第一设计更简单、更紧凑的筒设计,因为所述设计消除了完全分离接地传感器的空间要求和使生物样品移动至单独接地传感器所需的导管的额外长度。另外,显著减小了覆盖整个传感器电路所需的样品体积。关于图36-38讨论的本发明的实施方案被构造成将单个生物(例如,全血)样品维持在单个导管(干燥的试剂和/或对每个试验特异性的底物位于其中)中。关于图36-38讨论的构型与图27-29的构型之间的主要差异是,在图36-38的构型中,样品被维持在单个导管中且被在串联传感器上面推动并锁定或保持就位,且位于传感器上的试剂和/或底物被特别设计成通过被动扩散溶解在样品中或保留在固定化层内。在进行凝固分析的实施例中,级联途径的活化及其检测发生在紧密靠近传感器的高浓度区域处。溶解在样品中的试剂和/或底物保持在印制它们的传感器附近,因此消除试验之间的任何潜在交叉干扰。对于其中可能存在化学或物理干扰的任何两个试验(例如,凝固试验),这是多路技术的一个重要要素。如在图36中所示,本发明的一些实施方案涉及用于筒930的集成的接地传感器和单个导管设计,其包含具有连接部940(例如,t-连接部)的导管935。连接部940被构造成将单个导管传感器导管945与减压导管950(例如,排气道)分离。单个传感器导管945被构造成经过包含至少一个分析物检测电极(例如,aptt电极)的接地/传感器芯片955(如关于图18讨论的)的第一区域、包含参比电极的至少一部分的第二区域、和包含至少一个相同的或不同的分析物检测电极(例如,pt电极)的第三区域。根据本发明的方面,通过使用关于图4、7a、7b、7c和9讨论的布置中的一种或多种固定化试剂/底物,形成在单个导管传感器导管945内的分析物检测电极。筒930还可以包含定位在传感器导管945内的流体障碍机构960和从传感器导管945连通至腔体970的导管965(例如,排气道)。在该实施方案中,腔体970被构造为废物室(如关于图24讨论的)。但是,在替代实施方案中,导管965可以被构造成连通至废物导管(如关于图24讨论的)。如在图36和37中所示,在筒930的运行中,使用双向隔膜泵(如关于图24描述的)使流体或生物样品从入口或样品容纳室移动至导管935并穿过连接部940。减压导管950(例如,排气道)具有阻塞物或毛细管档栓(例如,毛细管爆裂阀或流体阻力/阻塞物)以使样品流转向至传感器导管945。将样品在接地/传感器芯片955上面推至流体障碍机构960(例如,在双面胶或模塑塑料部件之一中形成的膜“海绵阀”或微通道),所述流体障碍机构960将样品有效地锁定进传感器导管945中。此后可以开始在传感器导管945中的分析。样品的任何残余压力或移动然后将前进到减压导管950中。在优选的实施方案中,设计减压导管950中的阻塞物或毛细管档栓,使得它具有比传感器导管945中的流体障碍机构更低的压力阻力。如在图36和38中所示,在筒930的替代运行中,使用双向隔膜可以使流体或生物样品从入口或样品容纳室移动至导管935。此后将样品在接地/传感器芯片955上面推至流体障碍机构960(例如,在双面胶或模塑塑料部件之一中形成的膜“海绵阀”或微通道),所述流体障碍机构960将样品有效地锁定进传感器导管945中。此后可以开始传感器导管945中的分析。应当理解,用于筒930的集成的接地传感器和单个导管设计有利地提供了单个筒,其能够在单个导管内同时或先后进行两个独立测定(例如,应当理解所述测定可以是不同的或相同的,例如,pt和aptt、pt和pt、aptt和aptt等),而不需要将生物样品与试剂和/或底物混合。根据该实施方案的方面,筒930的部件允许在传感器导管945内进行两个单独分析试验,无需担忧在一种或多种试剂已经变得暴露于生物样品以后级联途径的交叉活化或其它交叉电极干扰,因为分析物检测电极是具有使用如本文中详细讨论的布置中的一种或多种形成的定位(例如,固定化)试剂/底物的微环境传感器。此外,用于筒930的集成的接地传感器设计提供了比上述的接地传感器第一设计更简单、更紧凑的筒设计,因为所述设计消除了完全分离接地传感器的空间要求和使生物样品移动至单独接地传感器所需的导管的额外长度。另外,显著减小了覆盖整个传感器电路所需的样品体积。关于图39讨论的本发明的实施方案被构造成分割单个生物(例如,全血)样品并允许在同一个一次性筒内在单个全血样品上同时或先后进行多个物理上分离的试验(例如,凝固试验和分析化学试验)。在某些实施方案中,为两个导管(干燥的试剂和/或对每个试验特异性的底物可以位于其中)中的至少两段样品提供独立混合控制。根据本发明的方面,所述底物可以是或不是定位(例如,固定化)在传感器上。溶解在样品中的试剂和/或底物保留在导管内且靠近印制或沉积它们的传感器,因此消除试验之间的任何潜在交叉干扰。对于其中可能存在化学或物理干扰的任何两个试验(例如,凝固试验和分析化学试验),这是多路技术的一个重要要素。如在图39中所示,本发明的一些实施方案涉及用于筒1000的多传感器构型,其包含具有连接部1010(例如,t-连接部)的导管1005。连接部1010可以包含第一阻塞物或毛细管档栓1015且被构造成将第一传感器导管1020与辅助导管1025分离。第一传感器导管1020被构造成经过包含至少一个分析物检测电极(例如,pt电极)的传感器芯片1030的至少一部分。根据本发明的方面,在有或没有使用关于图2、3、4、7a、7b、7c和9讨论的布置中的一种或多种定位(例如,固定化)试剂/底物的情况下,可以形成在第一传感器导管1020内的分析物检测电极。第二阻塞物或毛细管档栓1035可以定位在第一传感器导管1020内,且导管1040(例如,排气道)可以被构造成从第一传感器导管1020连通至废物导管1045(如关于图24讨论的)。筒1000还可以包含将腔体1055与废物导管1045连接的第二传感器导管1050。辅助导管1025在连接部1060处连接至第二传感器导管1050。连接部1060可以包含第三阻塞物或毛细管档栓1065。在某些实施方案中,第一阻塞物或毛细管档栓1015和第三阻塞物或毛细管档栓1065被构造成大于(例如,在宽度方面大于)第二阻塞物或毛细管档栓1035以允许如下文详细讨论的样品的控制。第二传感器导管1050被构造成经过一个或多个传感器芯片1070(其包含至少一个分析物检测电极(例如,钠或氯化物电极))中的每一个的至少一部分。一个或多个传感器芯片1070可以被构造成进行任何数目的测定,包括电解质、一般化学物质、血液气体和血液学(参见,例如,美国专利号7,419,821、6,379,883、5,514,253、5,200,051和5,096,669,它们通过引用整体并入本文)。例如,一个或多个传感器芯片1070可以被构造成进行任何数目的测定,所述测定能够检测一个或多个选自以下的分析物:氧分压、二氧化碳分压、总二氧化碳、ph、钾、钠、氯化物、葡萄糖、bun、肌酸酐、乳酸盐、镁、血细胞比容、离子化的钙、肌钙蛋白i、肌钙蛋白t、ckmb、原降钙素、bhcg、hcg、ntprobnp、probnp、bnp、肌红蛋白、甲状旁腺激素、d-二聚体、ngal、半乳糖凝集素-3和/或psa、以及其它分析物。在其中将底物用于进行测定的实施方案中,在有或没有使用关于图2、3、4、7a、7b、7c和9讨论的布置中的一种或多种定位(例如,固定化)试剂/底物的情况下,可以形成在第二传感器导管1050内的至少一个分析物检测电极。在不利用底物的其它实施方案中,可以在没有任何底物的情况下形成在第二传感器导管1050内的至少一个分析物检测电极。如在图39中所示,在筒1000的运行中,分析仪对垫圈的变形可以将压力传递到位于腔体1055中的装满流体(例如,大约130μl分析/洗涤溶液、对照流体或校正流体)的含流体的箔包上,使所述箔包破裂,并将流体驱逐进第二传感器导管1050中和经过第三阻塞物或毛细管档栓1065用于随后在样品分析过程中使用(如关于图24讨论的)。在某些实施方案中,所述箔包是含有校正溶液的校正包(calpak)。事件的典型顺序包括将calpak破裂,并然后使校准溶液在一个或多个传感器芯片1070上面经过以润湿一个或多个传感器芯片1070。此后,使用双向隔膜泵(如关于图24描述的)将流体或生物样品从入口或样品容纳室移动至导管1005。将生物样品在连接部1010(例如,t-连接部)处分成第一部分和第二部分。在优选的实施方案中,定位在辅助导管1025内的第一阻塞物或毛细管档栓1015(例如,毛细管爆裂阀或流体阻力/阻塞物)造成样品的第一部分优先填充第一传感器导管1020并在传感器芯片1030和至少一个电极(例如,pt电极)上面移动。在第一传感器导管1020内的样品的第一部分在第二阻塞物或毛细管档栓1035处停止,这会造成样品的第二部分挤过第一阻塞物或毛细管档栓1015和第三阻塞物或毛细管档栓1065。样品的第二部分填充第二传感器导管1050并在一个或多个传感器芯片1070和至少一个分析物检测电极(例如,钠或氯化物电极)上面移动。在某些实施方案中,样品的第二部分可以被构造成与存在于第二传感器导管1050内的分析/洗涤溶液、对照流体或校正流体混合。为了能够混合流体段,可以将部件(包括保留结构诸如立柱阵列(postarray)、导管切块(conduitcut-outs)、槽或微坑)设计进第二传感器导管1050中以保留分析/洗涤溶液、对照流体或校正流体。可替换地,通过在连接部1060处合并来自导管1025和箔包1055的两个流体流,可以完成两个流体段的混合。在其它实施方案中,分析/洗涤溶液、对照流体或校正液可以已经通过第二传感器导管1050泵送至废物导管1045,使得样品的第二部分不会与分析/洗涤溶液、对照流体或校正液混合。此外,为了使分析/洗涤溶液、对照流体或校正液的携带污染最小化,可以以在第一流体和样品的第二部分之间引入空气段的方式设计筒。辅助导管1025的体积将决定流体段之间的空气隙的大小。应当理解,用于筒1000的多传感器构型有利地提供了单个筒,其能够在两个单独导管内同时或先后进行两个独立测定(例如,pt和分析化学测定、pt和pt、aptt和pt、aptt和aptt等)。在其中需要混合或混合有利的实施方案中,筒1000的部件允许在第一导管和第二导管内的独立混合控制,无需担忧在一种或多种试剂已经变得暴露于生物样品以后的交叉活化或其它交叉电极干扰,因为所述传感器通过至少第一传感器导管和第二传感器导管的使用彼此物理上分离。如在图40(其中x-轴是时间/秒且y-轴是电流/pa)中所示,关于在筒的两个单独导管中的样品的独立混合控制(如在图25-29和39中所示)和充分混合试剂/底物(即,没有定位的微环境)的实施方案能够使用多个样品类型(由应答曲线1071代表的全血,由应答曲线1072和1073代表的除去因子的血液的2个水平,由应答曲线1075代表的对照血浆的1个水平)完成凝固结果(例如,由在每条凝固曲线上标记的垂直线指示的pt时间)。如在图41a中所示,关于不在筒的一个或多个导管中在固定化的或定位的试剂/底物上面混合样品的本发明的实施方案(如在图30-38中所示)也能够使用与图40中使用的那些样品类似的样品(由应答曲线1076代表的全血,由应答曲线1077代表的除去因子的血液,和由应答曲线1078和1079代表的对照血浆的2个水平)实现凝固结果。如在图41b中所示(由应答曲线1076代表的全血,由应答曲线1077代表的除去因子的血液,和由应答曲线1078和1079代表的对照血浆的2个水平),关于在固定化的或定位的试剂/底物上面混合样品的实施方案的传感器应答也是可实现的,但是,除去和延长因子的血浆对照样品的分辨率不像在固定化的无混合实施方案中那样清楚(如在图41a中所示)。进一步,优选的是,全血的凝固时间(曲线1071和1076)非常接近正常血浆对照凝固时间(曲线1072和1078);无混合的、固定化的/定位的实施方案反映这是最佳的,从而进一步证实它们相对于图40的系统的改进。尽管实施方案的无混合或混合形式是可能的,在实施方案的无混合形式(如在图41a中所示)中电流(pa)产生较高且变动性(pa和凝固时间)较低。这些意外的且更一致的结果可直接归因于如本文中关于无混合实施方案所述的微环境传感器的使用。与定位的(例如,固定化的)试剂/底物印制物组合的无混合实施方案代表较高试剂/底物与较低样品体积之比的凝固信号的活化/传播的系统改进,从而产生更快的测定/传感器应答时间。另外,直接在传感器上面的试剂/底物印制物的定位(例如,固定化)会导致扩散的底物离去基团一旦被活性凝血酶产生就立即周转(氧化)。这最终导致在电流测定传感器处直接产生更大的(和更可再现的)电流信号。最后,快速凝血酶应答(从图41a中的曲线的快速升高与图41b中的较慢升高的对比看出)与较高电流和较高再现性的组合会产生更容易地且再现地分析的应答曲线,从而产生与图40或41b中的那些测定中的任一种相比改进的测定(在图41a中表示的电流实施方案)。接地芯片消除和筒鉴别在优选的实施方案中,可以将接地芯片整合或集成进如本文中详细描述的传感器芯片中。一种典型的接地芯片(如关于图17描述的)可以包括充当传感器芯片的返回(return)的接地电极和用于筒鉴别的4个接触针(参见,例如,美国专利号7,419,821,其以它的整体并入本文)。因此,接地芯片在传感器芯片中的集成包括将这两种功能移入传感器芯片中。将接地芯片与传感器芯片集成的优点包括(i)简化的生产过程,因为在晶片制造、镀金属、划片和筒组装过程中少处理一个部件,(ii)降低的成本,和(iii)减小的样品体积,因为可以缩短传感器导管,如至少图25和31之间的对比所示。如在图42中所示,单独的接地芯片和传感器芯片布置1080(例如,图17中所示的布置)通常如下起作用:使用与接地芯片1090上的接地针1085和传感器芯片1100上的电流测定针1095连接的检测器1082来检测参比电极1105和分析物检测电极1110(例如,电流测定电极)之间的电流差异。为了将参比电极功能性赋予单个传感器芯片布置1115(例如,图18中所示的布置),通过将参比电极1105与电导测定低针1120连接可以将参比电极1105与传感器芯片1100集成。电子开关1125可以在分析仪中实现,所述分析仪被构造成连接接地针1085和电导测定低针1120。因此,参比电极1105可以基本上从接地芯片1090移动至传感器芯片1100。为了将筒鉴别功能性赋予单个传感器芯片布置(例如,图18中所示的布置),可以在用于筒鉴别的布置中包括另外的机构或装置。如在图42中关于单个电极布置1130(例如,aptt)所示,在未使用的电流测定针1140和电导测定低针1120之间仅可以实现分析物检测电极1110、电阻器1135。可以将分析物检测电极1110连接至电流测定针1095,可以将电阻器1135连接至未使用的电流测定针1140和电导测定低针1120,且可以将参比电极1105连接至电导测定低针1120。可以如下用检测器1145(例如,处理器)测量电阻器1135的电阻:在将筒插入分析仪中之后(例如,之后立即),在未使用的电流测定针1140和电导测定低针1120之间施加小电压,例如,1mv。测量的电阻的值然后可以用于筒鉴别。例如,每个筒类型(例如,筒ec8+、cg8+、eg7+、chem8+等)可以与某个电阻或电阻范围相关,使得所述筒的测量电阻可以用于使用查表法鉴别筒的类型。在某些实施方案中,电阻器1135可以由金属丝,优选与接触垫和传感器电极同时制造的金丝构成。金丝可以小至5μm宽和0.1μm厚,其形成0.5μm2的面积。由于金的电阻率是2.44μω-cm或0.0244ω-μm,1000μm长的金丝将具有以下电阻:0.0244ω-μm*1000μm/0.5μm2=48.8ω。将筒插入分析仪中以后,可以应用小电压,例如,0.5mv,且可以产生约10ua的电流并用分析仪检测。为了使功率消耗最小化,任选地所述金丝可以较长,施加的电压可以较低,或电压的施加时间可以较短。在一个替代实施方案中,单个电极布置1130可以包括仅pt的分析物检测电极1110,而不是仅aptt的分析物检测电极1110。根据本发明的该方面,金丝的长度可以增加至约10cm,这会使金丝的电阻增加至约5000ω,以便将pt筒的鉴别(identification)与aptt筒的鉴别区分开。在其它实施方案中,可以在电流测定针1095和电导测定低针1120之间应用电阻器。本领域普通技术人员应当理解,使用电阻器来鉴别筒的类型的概念可以在本文描述的任意传感器/筒布置中实现。此外,通过改变所述丝的几何形状或使用所述丝的不同材料(例如,使用tiw而不是金)可以得到电阻器的不同值,其然后可以用于鉴别不同的筒,而不脱离本发明的精神和范围。为了将筒鉴别功能性赋予多传感器芯片布置(例如,图18中所示的布置),可以在布置中包括用于筒鉴别的另外机构或装置。如在图43中关于多(例如,2)电极布置1150(例如,pt分析物检测电极1155和aptt分析物检测电极1160)所示,可以在未使用的电流测定针1170和电导测定低针1175之间应用电阻器1165。pt分析物检测电极1155可以连接至电流测定针1180,aptt分析物检测电极1160可以连接至电流测定针1185,电阻器1165可以连接至未使用的电流测定针1170和电导测定低针1175,且参比电极1190可以连接至电导测定低针1175。可以如下用检测器1195测量电阻器1165的电阻:在将筒插入分析仪中之后(例如,之后立即),在未使用的电流测定针1170和电导测定低针1175之间施加小电压,例如,1mv。测量的电阻的值然后可以用于筒鉴别。例如,每个筒类型(例如,筒ec8+、cg8+、eg7+、chem8+等)可以与某个电阻或电阻范围相关,使得所述筒的测量电阻可以用于使用查表法鉴别筒的类型。如以上所讨论的,电阻器1165可以由金属丝,优选与接触垫和传感器电极同时制造的金丝构成。金丝可以小至5μm宽和0.1μm厚,其形成0.5μm2的面积。由于金的电阻率是2.44μω-cm或0.0244ω-μm,1000μm长的金丝将具有以下电阻:0.0244ω-μm*1000μm/0.5μm2=48.8ω。将筒插入分析仪中以后,可以应用小电压,例如,0.5mv,且可以产生约10ua的电流并用分析仪检测。为了使功率消耗最小化,任选地所述金丝可以较长,施加的电压可以较低,或电压的施加时间可以较短。用在凝固测定中的鞣花酸制剂在优选的实施方案中,提供了包含鞣花酸制剂的凝固试验试剂,所述鞣花酸制剂包括鞣花酸和用于稳定地溶解所述鞣花酸的介质。所述鞣花酸是凝固活化剂且被构造成活化内源性途径,例如,以进行aptt测定,如本文中详细讨论的。所述介质是被构造成克服鞣花酸的水不溶解性和稳定地溶解鞣花酸的溶剂或化合物。所述介质可以包含氢氧化钠、甲醇、聚醚化合物和环糊精中的一种或多种。在一个实施方案中,所述介质可以包含氢氧化钠。氢氧化钠可以用作溶剂来帮助溶解不溶于水的、具有酸性官能团的化合物诸如鞣花酸。氢氧化钠与酸性官能团反应并形成水溶性的盐。在一个实施方案中,通过将鞣花酸与氢氧化钠和任选的去离子水混合以形成鞣花酸溶液,可以制备包含氢氧化钠作为用于溶解鞣花酸的溶剂的鞣花酸制剂。但是,基于醇基的脱水和/或在鞣花酸中存在的两个酯基的皂化反应,氢氧化钠向鞣花酸的添加会产生分解产物(即,无活性的鞣花酸)。结果,在某些实施方案中,可以将鞣花酸溶液进行ph中和以保留至少40%(优选地至少80%)的鞣花酸作为活性鞣花酸,例如,鞣花酸的两个酯基尚未经历皂化反应,这会增加鞣花酸溶液的稳定性。例如,鞣花酸制剂的制备还可以包含:(i)在中性ph将鞣花酸溶解在含有所述介质的溶液中,(ii)将碱(例如,氢氧化钠)加入所述溶液中以增加所述溶液的ph至10-12.5的ph范围,(iii)将所述溶液混合约1-10分钟的时间段,和(iv)将酸(例如,盐酸)加入所述活化剂溶液中以将ph恢复至中性ph(例如,约7.3的ph),从而保留至少40%(优选地至少80%)的鞣花酸作为活性鞣花酸(例如,未皂化的鞣花酸)。此后,根据本发明的不同方面,可以将鞣花酸溶液微分配为用在凝固测定中的试剂或试剂的组成部分。在另一个实施方案中,所述介质可以包含甲醇。甲醇是一种可与水和有机溶剂完全混溶的无色液体。已经证实鞣花酸微溶于甲醇。在一个实施方案中,通过将鞣花酸与甲醇和任选的去离子水混合以形成鞣花酸溶液,可以制备包含甲醇作为用于溶解鞣花酸的溶剂的鞣花酸制剂。在某些实施方案中,可以将鞣花酸溶液进行ph中和以增加鞣花酸溶液的稳定性。此后,根据本发明的不同方面,可以将鞣花酸溶液微分配为用在凝固测定中的试剂或试剂的组成部分。但是,甲醇溶解度通常不适合用于本文关于一次性使用的诊断筒描述的许多凝固测定实施方案。在另一个实施方案中,所述介质可以包含聚醚化合物。聚醚化合物是通过在许多较简单化合物(单体)的分子之间建立醚键而将它们连接在一起或聚合来制备的一类有机物质;聚醚,其在分子结构上可以是链样或网络样。聚醚化合物是在化妆品和药物制剂中和在乳化剂或润湿剂和润滑剂的制备中使用的通常水溶性的液体或蜡状固体。具体地,已经证实聚乙二醇(peg)具有高溶解度性质且经常用作溶剂和润滑剂(参见,例如,bala等人,2006,journalofpharmaceuticalandbiomedicalanalysis,40:206-10)。但是,以前没有探究peg用于溶解鞣花酸的用途及其在一次性使用的凝固测定装置中的性能。聚醚化合物可以选自peg、聚环氧乙烷、聚氧乙烯、及其混合物。在某些实施方案中,所述介质包含具有约200至约500000的分子量、更具体地约200-4000的分子量、优选地约200至约400的分子量的peg。在一个实施方案中,通过将鞣花酸与聚醚化合物和任选的去离子水混合以形成鞣花酸溶液,可以制备包含聚醚化合物(例如,peg)作为用于溶解鞣花酸的溶剂的鞣花酸制剂。此后,根据本发明的不同方面,可以将鞣花酸溶液微分配为用在凝固测定中的试剂或试剂的组成部分。在另一个实施方案中,所述介质可以包含环糊精。环糊精是一种环样分子,其可以如下增加溶解度:允许不溶性分子与疏水芯形成包含物(inclusion),同时仍然呈现更溶于水的亲水外部特征(raja&kumar,2013,journalofglobaltrendsinpharmaceuticalsciences,4(2):1099-1106)。存在几个制备环糊精包合络合物的方案(javery&doshi,2014,internationaljournalofuniversalpharmacyandbiosciences,3(3):674-91)。除了标准的环糊精以外,几个生产商已经制备了会提供增强的溶解度特征的衍生物(loftsson&brewster,2013,drugsolubilizationandstabilizationbycyclodextrindrugcarriersindrugdeliverystrategiesforpoorlywater-solubledrugs(douroumis等人编),wiley-blackwellpublisher,第67-101页)。但是,以前没有探究环糊精用于溶解鞣花酸的用途及其在一次性使用的凝固测定装置中的性能。环糊精可以选自(2-羟丙基)-β-环糊精、(2-羟丙基)-γ-环糊精、α-环糊精、γ-环糊精、二甲基-α-环糊精、三甲基-α-环糊精、二甲基-β-环糊精、三甲基-β-环糊精、二甲基-γ-环糊精、三甲基-γ-环糊精、糖基-α-环糊精、糖基-β-环糊精、二糖基-β-环糊精、麦芽糖基-β-环糊精、二麦芽糖基-β-环糊精、磺丁基醚-β-环糊精、及其混合物。在某些实施方案中,所述环糊精与鞣花酸络合。更具体地,所述环糊精与鞣花酸形成可溶性主体-客体复合物,具有至少1:1(鞣花酸:环糊精)的摩尔比,优选地选自1:1、1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9和1:10(鞣花酸:环糊精)的摩尔比。在一个实施方案中,通过将鞣花酸与环糊精和任选的去离子水混合以形成鞣花酸溶液,可以制备包含环糊精作为增加鞣花酸溶解度的化合物的鞣花酸制剂。在某些实施方案中,通过暴露于氢氧化物碱并用酸中和至中性ph,可以使鞣花酸溶液增溶。例如,通过加入氢氧化钠以增加ph(例如,约12的ph),且此后加入盐酸以降低ph至中性(例如,约7.3的ph),可以使鞣花酸溶液增溶。可以将鞣花酸溶液进行ph中和以保留至少40%(优选地至少80%)的鞣花酸作为活性鞣花酸(例如,未皂化的鞣花酸),这会增加鞣花酸溶液的稳定性。例如,鞣花酸制剂的制备还可以包含:(i)在中性ph将鞣花酸溶解在含有所述介质的溶液中,(ii)将碱(例如,氢氧化钠)加入所述溶液以将所述溶液的ph增加至10-12.5的ph范围,(iii)将所述溶液混合约1-10分钟的时间段,和(iv)将酸(例如,盐酸)加入所述活化剂溶液以将ph恢复至中性ph(例如,约7.3的ph)以保留至少40%(优选地至少60%)的鞣花酸作为未皂化的鞣花酸。此后,根据本发明的不同方面,可以将鞣花酸溶液微分配为用在凝固测定中的试剂或试剂的组成部分。在另一个实施方案中,所述介质可以是聚醚化合物和环糊精。可以如下制备包含聚醚化合物作为溶剂和环糊精作为增加鞣花酸溶解度的化合物的鞣花酸制剂:将环糊精与去离子水混合以形成环糊精的乳状液。将鞣花酸与聚醚化合物混合以形成鞣花酸溶液。将环糊精的乳状液与鞣花酸溶液混合,使得环糊精与鞣花酸络合以产生环糊精和鞣花酸乳状液。此后,根据本发明的不同方面,可以将环糊精和鞣花酸乳状液微分配为用在凝固测定中的试剂或试剂的组成部分。在某些实施方案中,所述凝固试验试剂可以包括促进所述试剂的稳定和/或沉积/溶解特征的另外凝固活化剂和/或多种其它组分。在某些实施方案中,所述另外凝固活化剂可以选自c盐(celite)、高岭土、硅藻土、粘土、二氧化硅、合成的或天然的脂质和合成的或天然的磷脂。在某些实施方案中,所述其它组分可以选自葡聚糖、环糊精、糊精、tergitol、缓冲剂、载体蛋白、一个或多个氨基酸、稳定剂、抗微生物剂、抗氧化剂、去污剂、糖化物(saccharide)、多糖、蔗糖、聚醚化合物诸如聚乙二醇、聚乙二醇衍生物、聚环氧乙烷或聚氧乙烯、甘氨酸、明胶、缓冲剂诸如4-(2-羟基乙基)-1-哌嗪乙磺酸(hepes)缓冲剂、鼠李糖、海藻糖、水和糖(sugar)。在优选的实施方案中,提供了凝固试验系统,其包含(i)试剂,其包含鞣花酸制剂(包括鞣花酸和用于稳定地溶解所述鞣花酸的介质)、磷脂和缓冲剂,和(2)底物,其包含一种或多种具有可检测部分的、凝血酶可切割的肽。所述磷脂可以选自1,2-二油酰基-sn-甘油-3磷酸胆碱(pc)、1,2-二油酰基-sn-甘油-3-磷酸乙醇胺(pe)、1,2-二油酰基-sn-甘油-3-磷酸肌醇(铵盐)(pi)和1,2-二油酰基-sn-甘油-3-磷酸-l-丝氨酸(钠盐)(ps)。所述缓冲剂可以选自4-(2-羟基乙基)-1-哌嗪乙磺酸(hepes)缓冲剂、n-2-羟基乙基哌嗪-n-2-乙磺酸(hepes)、3-(n-吗啉代)-丙磺酸(mops)、tris、n,n-二-(羟乙基)-2-氨基乙磺酸(bes)、n-三-(羟甲基)-甲基-2-氨基乙磺酸(tes)、3-[n-三(羟基甲基)甲基氨基]-2-羟基丙磺酸(tapso)和3-[n-三-(羟基甲基-甲基氨基]-丙磺酸(taps)。如本文所讨论的,所述底物可以具有酰胺键,其模仿凝血酶切割的纤维蛋白原中的酰胺键。具体地,所述底物可以包含一种或多种凝血酶可切割的肽,诸如选自以下的那些:h-d-phe-pip-arg、h-d-chg-abu-arg、cbz-gly-pro-arg、boc-val-pro-arg、h-d-phe-pro-arg、环己基甘氨酸-ala-arg、tos-gly-pro-arg、bz-phe-val-arg、boc-val-pro-arg、ac-val-pro-arg、ac-val-hyp-arg、ac-(8-氨基-3,6-二氧杂辛酰基-val-pro-arg、ac-gly-pro-arg、ac-(8-氨基-3,6-二氧杂辛酰基-gly-pro-arg、ac-gly-hyp-arg和h-d-chg-abu-arg。凝血酶通常在精氨酸残基的羧基端处切割酰胺键,因为所述键在结构上类似于凝血酶切割的纤维蛋白原中的酰胺键。凝血酶-底物反应的产物包括电化学上惰性的化合物诸如tos-gly-pro-arg、h-d-phe-pip-arg和/或bz-phe-val-arg-和电活性的化合物或可检测的部分,优选地选自对-氨基苯酚、醌、二茂铁、亚铁氰化物衍生物、其它有机金属物质、对-硝基苯胺、邻-二茴香胺、4,4’-联苯胺、4-甲氧基-2-萘基胺、n-苯基-对苯二胺、n-[对甲氧基苯基-]-对苯二胺和吩嗪衍生物。在优选的实施方案中,所述凝固试验系统还可以包含至少一个转换器(例如,工作电极或光学检测器)。在某些实施方案中,所述试剂和底物可以在至少一个转换器的表面附近以定位方式形成。例如,所述试剂和所述底物可以作为干燥物质直接印制在至少一个转换器的表面上。流体样品可以在没有混合(例如,通过被动扩散)的情况下以定位方式与所述试剂和所述底物反应反应(尽管可能期望某种程度的混合,例如,流体振荡),从而建立从传感器至导管顶部的活性凝血酶(来自鞣花酸对凝固途径的活化)梯度,且所述凝血酶将底物和纤维蛋白原同时转化成它们的切割产物。由至少一个转换器检测电活性的化合物或可检测部分。在某些实施方案中,所述凝固试验系统的至少一个转换器可以是微制造的传感器阵列的一部分。例如,微制造的传感器阵列可以包含一个或多个传感器或转换器,其包含第一传感器或转换器(例如,aptt传感器)和任选的第二传感器或转换器(例如,pt传感器)。为了便利传感器阵列内的凝固试验系统的运行,可以将凝固试验系统构建为微环境传感器结构,其包含处于许多不同布置中的任一种的试剂和底物,使得流体样品(例如,全血)向试剂和底物的引入定位至至少一个转换器。如本文中讨论的。具体地,微环境传感器结构被构造成使一种或多种试剂和/或反应产物物理上分离以在一种或多种试剂已经变得暴露于流体样品以后避免级联途径的交叉活化或其它传感器交叉干扰。为了将凝固试验系统与其它测定物理上分离以避免交叉活化和促进电化学或光信号在转换器上的定位,凝固试验系统还可以包含选择性地模印在至少转换器上面的固定化的底物和/或试剂-聚合物层。例如,包含试剂、底物和聚合物(诸如可光变形的聚合物)中的一种或多种的水性聚合物基质可以用于将试剂和/或底物以许多不同布置中的任一种固定化在至少一个转换器上或附近,如本文中讨论的(参见,例如,图7a-7c和13a-13c)。在某些实施方案中,所述聚合物可以选自羟丙基纤维素和elvace、羧甲基纤维素、聚乳酸、聚乳酸、聚乳酸-乙醇酸共聚物、纤维素、纤维素衍生物、羟丙基甲基醋酸纤维素琥珀酸酯、菊糖、果糖、果聚糖、果聚糖衍生物和聚乙醇酸。另外,所述聚合物层还可以包含。有利地,本发明的鞣花酸制剂被构造成以用在凝固试验溶液中的高可溶形式提供鞣花酸。甚至更有利地,本发明的鞣花酸制剂被构造成以对于长期储存而言非常稳定且减小aptt凝固测定的测定时间的高可溶形式提供鞣花酸。考虑到下述非限制性实施例将更好地理解本发明。实施例实施例1:在氢氧化钠中的鞣花酸最初,将0.0204g来自sigma(目录号e2250-10g)的鞣花酸加入300μl的1mnaoh中以产生0.34%(3.4mg/ml)部分地溶解的鞣花酸溶液。此后,将300μl部分地溶解的鞣花酸溶液用5.7ml去离子水稀释以产生稀释的溶解的鞣花酸溶液。此后,将1ml稀释的溶解的鞣花酸溶液用33ml去离子水进一步稀释以产生大约100μg/ml的稀释的溶解的鞣花酸溶液用于高效液相色谱法(hplc)注射。保留稀释的溶解的鞣花酸溶液的样品用于新鲜hplc注射,并将稀释的溶解的鞣花酸溶液的另一个样品在hplc注射之前在室温放置储存2天。hplc条件如下:使用agilentzorbaxeclipsexdb-c18柱(5um,4.6×150mm)以0.6ml/min的流速在agilent1200hplc仪器上色谱分离鞣花酸制品。流动相是使用下述梯度表1的a:乙腈,和b:1%乙酸。表1:时间(min)a(%)b(%)059835158555257573554578100083100090595图44-57显示了在260nm波长处捕获的峰强度(mau)相对于保留时间的图,但是使用其它波长(例如,280、320和360nm)确定任意其它污染物的存在。从每个运行捕获峰面积百分比和强度。图44显示了在氢氧化钠中新鲜制备的鞣花酸制品的hplc色谱图。图45显示了在氢氧化钠中制备并在室温储存2天的鞣花酸制品的hplc色谱图。来自图44和45的数据表明,在氢氧化钠中制备的鞣花酸正在经历分解(即,从92.9%下降至66.7%)。可以预期,就观察到的分解速率而言,皂化反应将依赖于时间、温度和浓度。该数据还表明,在没有ph中和下在有氢氧化钠存在下鞣花酸的储存将在凝固测定(诸如aptt测定)中产生可变的结果。实施例2:在甲醇中的鞣花酸最初,将0.0364g来自sigma(目录号e2250-10g)的鞣花酸加入3.64ml甲醇中从而产生具有约10mg/ml的浓度的部分地溶解的鞣花酸溶液。迅速地混合部分地溶解的鞣花酸溶液以后,将100μl的10mg/ml溶液加入9.9ml甲醇中,从而产生大约100μg/ml的溶解的鞣花酸溶液用于hplc注射。保留溶解的鞣花酸溶液的样品用于新鲜hplc注射,将溶解的鞣花酸溶液的另一个样品在hplc注射之前在室温放置储存6天,并将溶解的鞣花酸溶液的另一个样品在hplc注射之前在室温放置储存13天。使用与实施例1关于讨论的相同hplc条件,将溶解的鞣花酸溶液的每个样品注射进hplc中。图46显示了在甲醇中新鲜制备的鞣花酸制品的hplc色谱图。图47显示了在甲醇中制备并在室温储存6天的鞣花酸制品的hplc色谱图。图48显示了在甲醇中制备并在室温储存13天的鞣花酸制品的hplc色谱图。来自图46-48的数据表明鞣花酸制品中没有显著的可观察的分解。实施例3:在1mpeg400中的鞣花酸最初,将0.0224g来自sigma(目录号e2250-10g)的鞣花酸加入6ml的1m的来自sigma(目录号91893-250ml-f)的peg400中,从而产生部分地溶解的鞣花酸溶液。将鞣花酸粉末快速地溶解在溶剂中。此后,将部分地溶解的鞣花酸溶液通过反转试管约10次进行混合,并用微尖探头声处理10分钟以产生溶解的鞣花酸溶液。保留溶解的鞣花酸溶液的样品用于新鲜hplc注射,将溶解的鞣花酸溶液的另一个样品在hplc注射之前在室温放置储存6天,并将溶解的鞣花酸溶液的另一个样品在hplc注射之前在室温放置储存13天。对于新鲜hplc注射,将27μl的溶解的鞣花酸溶液用1ml去离子水稀释,从而产生大约100μg/ml的稀释的溶解的鞣花酸溶液用于hplc注射。对于在室温储存6天和13天的hplc注射,将27μl的每种溶解的鞣花酸溶液用1ml甲醇稀释,从而产生大约100μg/ml的稀释的溶解的鞣花酸溶液用于hplc注射。使用与实施例1关于讨论的相同hplc条件,将溶解的鞣花酸溶液的每个样品注射进hplc中。图49显示了在1mpeg400中制备并在hplc注射之前在去离子水中稀释的新鲜鞣花酸制品的hplc色谱图。图50显示了在1mpeg400中制备、在室温储存6天、并在hplc注射之前在甲醇中稀释的鞣花酸制品的hplc色谱图。图51显示了在1mpeg400中制备、在室温储存13天、并在hplc注射之前在甲醇中稀释的鞣花酸制品。来自图49-51的数据提示,当用1mpeg400制备时,所述鞣花酸制品在室温稳定13天。实施例4:在100mmpeg4000中的鞣花酸最初,将20gpeg4000(sigma95904-250g-f)加入去离子水中直到50ml以产生100mmpeg4000溶液,将其在旋转器上混合1小时。此后,将0.0494g来自sigma(目录号e2250-10g)的鞣花酸加入6ml的100mmpeg4000溶液中,从而产生鞣花酸和peg的不溶性混合物。将另外13.8ml的100mmpeg4000溶液加入鞣花酸和peg的不溶性混合物中以产生具有大约0.34%鞣花酸浓度的混合物,将其在旋转器上混合过夜。此后,将部分地溶解的鞣花酸溶液在50%用微尖探头声处理10分钟以产生溶解的鞣花酸溶液。保留溶解的鞣花酸溶液的样品用于新鲜hplc注射,将溶解的鞣花酸溶液的另一个样品在hplc注射之前在室温放置储存6天,并将溶解的鞣花酸溶液的另一个样品在hplc注射之前在室温放置储存13天。对于新鲜hplc注射和在室温储存6天和13天的hplc注射,将29.4μl的每种溶解的鞣花酸溶液用1ml甲醇稀释,从而产生大约100μg/ml的稀释的溶解的鞣花酸溶液用于hplc注射。使用与实施例1关于讨论的相同hplc条件,将溶解的鞣花酸溶液的每个样品注射进hplc中。图52显示了在100mmpeg4000中制备并在室温储存6天的鞣花酸制品的hplc色谱图,其与新鲜制品(数据未显示)相比没有显著变化。图53显示了在100mmpeg4000中制备并在室温储存13天的鞣花酸制品。来自图52和53的数据提示,当用100mmpeg4000制备时,所述鞣花酸制品在室温稳定13天。实施例5:在peg400和β-环糊精中的鞣花酸最初,将0.6614g来自sigma(目录号c4805-100g)的β-环糊精加入6.0ml去离子水中,从而产生β-环糊精的乳状液。此后,将0.3003g来自sigma(目录号e2250-10g)的鞣花酸加入8ml的1m的peg400溶液中并涡旋以产生鞣花酸的浓乳状液。高速搅拌鞣花酸的浓乳状液以后,将2ml的β-环糊精乳状液加入鞣花酸的浓乳状液中。此后,将β-环糊精和鞣花酸乳状液在室温搅拌1小时,并用微尖探头声处理10分钟以产生混合的β-环糊精和鞣花酸乳状液。将混合的β-环糊精和鞣花酸乳状液在室温静置过夜。静置过夜以后,混合的β-环糊精和鞣花酸乳状液不具有可观察的沉淀物。为了产生(1×)浓度的混合的β-环糊精和鞣花酸乳状液,将1ml的混合的β-环糊精和鞣花酸乳状液加入9ml的1m的peg400溶液中。此后,将33.3μl的(1×)浓度的混合的β-环糊精和鞣花酸乳状液加入1ml甲醇中以产生大约100μg/ml的稀释的溶解的鞣花酸溶液用于hplc注射。保留溶解的鞣花酸溶液的样品用于新鲜hplc注射,并将溶解的鞣花酸溶液的另一个样品在hplc注射之前在室温放置储存6天。使用与实施例1关于讨论的相同hplc条件,将溶解的鞣花酸溶液的每个样品注射进hplc中。图54显示了用β-环糊精制备的新鲜鞣花酸制品的hplc色谱图。图55显示了用β-环糊精制备并在室温储存6天的鞣花酸制品的hplc色谱图。来自图56和57的数据表明用β-环糊精制备的鞣花酸制品没有显著的可观察的分解。实施例6:在ph休克处理的β-环糊精中的鞣花酸最初,将0.3454g来自sigma(目录号e2250-10g)的鞣花酸加入2.65g来自sigma(目录号c4805-100g)的β-环糊精中并混合以产生鞣花酸的乳状液。此后,将β-环糊精和鞣花酸乳状液用5ml去离子水稀释并在高凝结(highsetting)下搅拌以产生稀释的β-环糊精和鞣花酸乳状液。此后,将10m氢氧化钠(约2-3ml)逐滴加入稀释的β-环糊精和鞣花酸乳状液中以产生看起来溶解的淡棕色模糊溶液。此后,将浓盐酸逐滴加入溶解的β-环糊精和鞣花酸乳状液中,同时测量溶液的ph以产生亮黄色β-环糊精和鞣花酸乳状液,其具有7.31的ph和11.5ml的终体积。为了产生(1×)浓度的混合的β-环糊精和鞣花酸乳状液,将1ml的混合的β-环糊精和鞣花酸乳状液加入9ml的1m的peg400溶液并声处理10分钟。此后,将29μl的(1×)浓度的混合的β-环糊精和鞣花酸乳状液加入1ml甲醇以产生大约100μg/ml的稀释的溶解的鞣花酸溶液用于hplc注射。保留溶解的鞣花酸溶液的样品用于新鲜hplc注射,并将溶解的鞣花酸溶液的另一个样品在hplc注射之前在室温放置储存6天。使用与实施例1关于讨论的相同hplc条件,将溶解的鞣花酸溶液的每个样品注射进hplc中。图56显示了用ph休克处理的β-环糊精制备的新鲜鞣花酸制品的hplc色谱图。图57显示了用ph休克处理的β-环糊精制备并在室温储存6天的鞣花酸制品的hplc色谱图。来自图56和57的数据表明在用ph休克处理的β-环糊精制备的鞣花酸制品中没有显著的可观察的分解。实施例7:aptt凝固混合液制备方法关于用作aptt试剂试验的样品包括:实施例1:1m氢氧化钠溶解的鞣花酸溶液,实施例3:1mpeg400溶解的鞣花酸溶液,实施例4:100mmpeg4000溶解的鞣花酸溶液,实施例5:peg400和β-环糊精溶解的鞣花酸溶液,和实施例6:ph休克处理的β-环糊精溶解的鞣花酸溶液。如下制备aptt混合液:最初,将0.06g来自sigma(目录号81323)的聚(乙二醇)甲基醚和30μl盐(1mnacl或kcl)加入一定比例的6ml的每种上述试验样品(实施例1、3、4、5和6)中以产生活化剂溶液。此后,将30g来自sigma(目录号c2432)的氯仿加入2个管形瓶中,每个管形瓶含有0.006g来自sigma(目录号b1253)的丁羟茴香醚。将磷脂1,2-二油酰基-sn-甘油-3磷酸胆碱(pc)、1,2-二油酰基-sn-甘油-3-磷酸乙醇胺(pe)、1,2-二油酰基-sn-甘油-3-磷酸肌醇(铵盐)(pi)和1,2-二油酰基-sn-甘油-3-磷酸-l-丝氨酸(钠盐)(ps)溶解在氯仿/丁羟茴香醚溶剂中。以磷脂1.16μl10.3mmps、1.69μl10.6mmpc、0.535μl11.2mmpe和17.5μl0.103mmpi的比例将它们混合。另外,用50mm来自sigma(目录号h0887-100ml)的hepes和0.5%的来自sigma(目录号r3875)的鼠李糖制备hepes/鼠李糖溶液。通过将75μl上面制备和试验的活化剂溶液(实施例1、3、4、5和6)加入20.9μl磷脂制品中,制备aptt“试剂”。aptt“底物”rf16-1具有tos-gly-pro-arg-meopada的序列。通过将20mg底物悬浮在7.366ml去离子水或10mm乙酸中以制备具有4mm终浓度的底物,制备aptt“底物”。如下制备溶液基质:将0.4288g来自sigma(目录号a-7906)的蛋清级分v加入2.538g来自sigma(目录号g-7126)的甘氨酸、0.3938g来自sigma(目录号81323)的甲氧基聚乙二醇和1.4g来自sigma(目录号s9378)的蔗糖中以产生溶液基质,用去离子水补至250ml。通过将0.065g来自aldrich(目录号435007)的羟丙基纤维素加入1ml去离子水中,制备6.5%的hpc(80000)溶液。如下制备用于微分配在前述凝固芯片上的aptt混合液:对于150μl的总体积,取30μl的aptt“试剂”,加入33μl的hepes/鼠李糖溶液、22μl的溶液基质、40μl的6.5%hpc(80000)、25μl的4mmaptt“底物”rf16-1。将该物质微分配在aptt芯片上并组装进筒装置中用于试验。图58显示了使用从含有全血的上述试验样品产生的aptt测定试剂得到的电流相对于时间的计时电流测定图(图a.用氢氧化钠制备的鞣花酸(基线)。图b.在peg4000溶液中制备的鞣花酸。图c.在peg400溶液中制备的鞣花酸。图d.在含有β-环糊精的peg400溶液中制备的鞣花酸。图e.在ph休克处理的β-环糊精溶液中制备的鞣花酸。为每个图指示了微分配的印制物的印制厚度)。稳定化的鞣花酸溶液能够产生与基线鞣花酸制品方法类似的凝固曲线和时间。证实了稳定的鞣花酸的老化制品会产生可接受的凝固参数(数据未显示)。实施例8:用于ph休克处理的鞣花酸的aptt凝固混合液制备方法如下制备aptt混合液:最初,将05ml去离子水放入50ml离心管中并加入0.3456g鞣花酸(sigmae2250-10g批号slbd8693v)以产生鞣花酸乳状液。将鞣花酸乳状液通过涡旋10秒进行混合。此后,将2.7ml的2mnaoh(每次加入以后在混合时一次加入675μl)加入鞣花酸乳状液中。此后,将3.8ml的1nhcl(每次加入以后在混合时一次加入950μl)加入鞣花酸乳状液中。记录鞣花酸乳状液的最终重量为13g,最终的ph为7.57。然后将鞣花酸乳状液在50%声处理10分钟,选择微探头选项。将该混合的鞣花酸乳状液标记为ea储备溶液。如下制备(1×)浓度的ea储备溶液:将0.5gea储备溶液称量进5mleppendorf试管中,向其中加入150μl的1mkcl/1%mepeg溶液和4350μl去离子水。此后将(1×)浓度的ea储备溶液用作活化剂溶液。通过将75μl上面制备的活化剂溶液加入75μl磷脂制品中,制备aptt“试剂”。以与关于实施例7讨论的方式类似的方式制备磷脂制品,且包括将20.9μl的ps:pc:pe:pi掺合物等分进玻璃瓶中,在温和氮气流下蒸发出氯仿,随后在50mmhepes+0.5%鼠李糖缓冲液中再水合。aptt“底物”rf16-1具有tos-gly-pro-arg-meopada的序列。如下制备aptt“底物”:将20mg底物悬浮于14.732ml去离子水中以制备具有2mm终浓度的底物。如下制备溶液基质:将0.4288g来自sigma(目录号a-7906)的蛋清级分v加入2.538g来自sigma(目录号g-7126)的甘氨酸、0.3938g来自sigma(目录号81323)的甲氧基聚乙二醇和1.4g来自sigma(目录号s9378)的蔗糖中以产生溶液基质,用去离子水补至250ml。通过将0.065g来自aldrich(目录号435007)的羟丙基纤维素加入1ml去离子水中,制备6.5%的hpc(80000)溶液。如下制备用于微分配在前述凝固芯片上的aptt混合液:取75μl的aptt“试剂”,加入6.6μl的1mhepes、13.4μl的40%鼠李糖溶液、55μl的溶液基质、100μl的6.5%hpc(80000)、125μl的2mmaptt“底物”rf16-1至375μl的总体积。将该物质微分配到aptt芯片上并装配进筒装置中用于试验。图59显示了使用aptt测定试剂和全血得到的电流相对于时间的计时电流测定图(a.通过ph休克方法制备的仅鞣花酸溶液)。实施例9:关于在1mpeg400中的鞣花酸的aptt凝固混合液制备方法如下制备aptt混合液:最初,将0.0224g来自sigma(目录号e2250-10g)的鞣花酸加入6ml的1m的来自sigma(目录号91893-250ml-f)的peg400中。将鞣花酸粉末快速地溶解在溶剂中。此后,将部分地溶解的鞣花酸溶液通过反转试管约10次进行混合,并用微尖探头声处理10分钟以产生溶解的鞣花酸溶液。为了产生(1×)浓度的溶解的鞣花酸溶液,将0.5ml溶解的鞣花酸溶液加入4.5ml去离子水中。此后,将30μl的1m的来自fisherscientific(目录号p-217-10)的kcl和0.01g来自sigma(目录号81323)的mepeg加入1ml(1×)溶解的鞣花酸溶液中。此后将该溶液用作活化剂溶液。通过将75μl上面制备的活化剂溶液加入75μl磷脂制品中,制备aptt“试剂”。以与关于实施例7讨论的方式类似的方式制备磷脂制品,且包括将20.9μl的ps:pc:pe:pi掺合物等分进玻璃瓶中,在温和氮气流下蒸发出氯仿,随后在50mmhepes+0.5%鼠李糖缓冲液中再水合。aptt“底物”rf16-1具有tos-gly-pro-arg-meopada的序列。如下制备aptt“底物”:将20mg底物悬浮在14.732ml去离子水中以制备具有2mm的终浓度的底物。如下制备溶液基质:将0.4288g来自sigma(目录号a-7906)的蛋清级分v加入2.538g来自sigma(目录号g-7126)的甘氨酸、0.3938g来自sigma(目录号81323)的甲氧基聚乙二醇和1.4g来自sigma(目录号s9378)的蔗糖中以产生溶液基质,用去离子水补至250ml。通过将0.065g来自aldrich(目录号435007)的羟丙基纤维素加入1ml去离子水中,制备6.5%的hpc(80000)溶液。如下制备用于微分配在前述凝固芯片上的aptt混合液:对于375μl的总体积,取75μl的aptt“试剂”,加入6.6μl的1mhepes、13.4μl的40%鼠李糖溶液、55μl的溶液基质、100μl的6.5%hpc(80000)、125μl的2mmaptt“底物”rf16-1。将该物质微分配到aptt芯片上并装配进筒装置中用于试验。图59显示了使用aptt测定试剂在全血中得到的电流相对于时间的计时电流测定图(b.在1mpeg400溶液中制备的鞣花酸)。实施例10:关于鞣花酸和β-环糊精的aptt凝固混合液制备方法如下制备aptt混合液:最初,在含有搅拌棒的30ml烧杯中,将0.3454g来自sigma(目录号e2250-10g)的鞣花酸加入2.65g来自sigma(目录号c4805-100g)的β-环糊精中。此后,将5.7ml去离子水和300μl的1m氢氧化钠加入β-环糊精和鞣花酸乳状液中并在高凝结下搅拌以产生溶解的β-环糊精和鞣花酸乳状液。此后,将500μl的2m氢氧化钠加入溶解的β-环糊精和鞣花酸乳状液中,同时测量溶液的ph以产生具有7.5ml终体积的亮黄色β-环糊精和鞣花酸乳状液。为了产生(1×)浓度的混合的β-环糊精和鞣花酸乳状液,将0.5ml混合的β-环糊精和鞣花酸乳状液加入4.5ml去离子水中并声处理10分钟。此后,将30μl的1m的来自fisherscientific(目录号p-217-10)的kcl和0.01g来自sigma(目录号81323)的mepeg加入1ml(1×)溶解的鞣花酸溶液中。此后将该溶液用作活化剂溶液。通过将75μl上面制备的活化剂溶液加入75μl磷脂制品中,制备aptt“试剂”。以与关于实施例7讨论的方式类似的方式制备磷脂制品,且包括将20.9μl的ps:pc:pe:pi掺合物等分进玻璃瓶中,在温和氮气流下蒸发出氯仿,随后在50mmhepes+0.5%鼠李糖缓冲液中再水合。aptt“底物”rf16-1具有tos-gly-pro-arg-meopada的序列。如下制备aptt“底物”:将20mg底物悬浮在14.732ml去离子水中以制备具有2mm的终浓度的底物。如下制备溶液基质:将0.4288g来自sigma(目录号a-7906)的蛋清级分v加入2.538g来自sigma(目录号g-7126)的甘氨酸、0.3938g来自sigma(目录号81323)的甲氧基聚乙二醇和1.4g来自sigma(目录号s9378)的蔗糖中以产生溶液基质,用去离子水补至250ml。通过将0.065g来自aldrich(目录号435007)的羟丙基纤维素加入1ml去离子水中,制备6.5%的hpc(80000)溶液。如下制备用于微分配在前述凝固芯片上的aptt混合液:对于375μl的总体积,取75μl的aptt“试剂”,加入6.6μl的1mhepes、13.4μl的40%鼠李糖溶液、55μl的溶液基质、100μl的6.5%hpc(80000)、125μl的2mmaptt“底物”rf16-1。将该物质微分配到aptt芯片上并装配进筒装置中用于试验。图59显示了使用aptt测定试剂在全血中得到的电流相对于时间的计时电流测定图(c.在含有β-环糊精的去离子水中制备的鞣花酸)。实施例11:关于ph休克处理的鞣花酸、peg和β-环糊精的aptt凝固混合液制备方法如下制备aptt混合液:最初,将2.65g来自sigma(目录号c4805-100g)的β-环糊精加入5.0ml去离子水和0.3454g来自sigma(目录号e2250-10g)的鞣花酸中,从而产生鞣花酸和β-环糊精的乳状液。此后,将2m氢氧化钠逐滴加入鞣花酸和β-环糊精乳状液中直到达到12的ph(大约2.5ml)。此后,将1m盐酸逐滴加入鞣花酸和β-环糊精乳状液中直到达到7.31的ph(大约3.7ml)。然后将鞣花酸和β-环糊精乳状液倒入50ml离心试管中并声处理10分钟。此后,将1.5ml去离子水加入鞣花酸和β-环糊精乳状液中以达到具有11.5ml的终体积的鞣花酸和β-环糊精乳状液。为了产生(1×)浓度的鞣花酸和β-环糊精乳状液,将0.2ml鞣花酸和β-环糊精乳状液加入1.8ml的50mmpeg400溶液中。此后,将30μl的1m的来自fisherscientific(目录号p-217-10)的kcl和0.01g来自sigma(目录号81323)的mepeg加入1ml(1×)溶解的鞣花酸溶液中。此后将该溶液用作活化剂溶液。通过将75μl上面制备的活化剂溶液加入75μl磷脂制品中,制备aptt“试剂”。以与关于实施例7讨论的方式类似的方式制备磷脂制品,且包括将20.9μl的ps:pc:pe:pi掺合物等分进玻璃瓶中,在温和氮气流下蒸发出氯仿,随后在50mmhepes+0.5%鼠李糖缓冲液中再水合。aptt“底物”rf16-1具有tos-gly-pro-arg-meopada的序列。如下制备aptt“底物”:将20mg底物悬浮在14.732ml去离子水中以制备具有2mm的终浓度的底物。如下制备溶液基质:将0.4288g来自sigma(目录号a-7906)的蛋清级分v加入2.538g来自sigma(目录号g-7126)的甘氨酸、0.3938g来自sigma(目录号81323)的甲氧基聚乙二醇和1.4g来自sigma(目录号s9378)的蔗糖中以产生溶液基质,用去离子水补至250ml。通过将0.065g来自aldrich(目录号435007)的羟丙基纤维素加入1ml去离子水中,制备6.5%的hpc(80000)溶液。如下制备用于微分配在前述凝固芯片上的aptt混合液:对于375μl的总体积,取75μl的aptt“试剂”,加入6.6μl的1mhepes、13.4μl的40%鼠李糖溶液、55μl的溶液基质、100μl的6.5%hpc(80000)、125μl的2mmaptt“底物”rf16-1。将该物质微分配到aptt芯片上并装配进筒装置中用于试验。图59显示了使用aptt测定试剂在全血中得到的电流相对于时间的计时电流测定图(d.在含有β-环糊精的50mmpeg400溶液中制备的鞣花酸)。实施例12:关于鞣花酸和甲基-环糊精的aptt凝固混合液制备方法如下制备aptt混合液:最初,将3.02g来自sigma(目录号c4555-10g)的甲基-环糊精加入5.0ml去离子水和0.3454g来自sigma(目录号e2250-10g)的鞣花酸中,从而产生鞣花酸和甲基-环糊精的乳状液。此后,将2m氢氧化钠逐滴加入鞣花酸和甲基-环糊精乳状液中直到达到12.5的ph。此后,将1m盐酸逐滴加入鞣花酸和甲基-环糊精乳状液中直到达到7.3的ph。然后将鞣花酸和甲基-环糊精乳状液倒入50ml离心试管中并声处理10分钟。此后,将1.5ml去离子水加入鞣花酸和甲基-环糊精乳状液中以达到具有11.5ml的终体积的鞣花酸和甲基-环糊精乳状液。为了产生(1×)浓度的鞣花酸和甲基-环糊精乳状液,将0.5ml鞣花酸和甲基-环糊精乳状液加入4.5ml去离子水中。此后,将30μl的1m的来自fisherscientific(目录号p-217-10)的kcl和0.01g来自sigma(目录号81323)的mepeg加入1ml(1×)溶解的鞣花酸溶液中。此后将该溶液用作活化剂溶液。通过将75μl上面制备的活化剂溶液加入75μl磷脂制品中,制备aptt“试剂”。以与关于实施例7讨论的方式类似的方式制备磷脂制品,且包括将20.9μl的ps:pc:pe:pi掺合物等分进玻璃瓶中,在温和氮气流下蒸发出氯仿,随后在50mmhepes+0.5%鼠李糖缓冲液中再水合。aptt“底物”rf16-1具有tos-gly-pro-arg-meopada的序列。如下制备aptt“底物”:将20mg底物悬浮在14.732ml去离子水中以制备具有2mm的终浓度的底物。如下制备溶液基质:将0.4288g来自sigma(目录号a-7906)的蛋清级分v加入2.538g来自sigma(目录号g-7126)的甘氨酸、0.3938g来自sigma(目录号81323)的甲氧基聚乙二醇和1.4g来自sigma(目录号s9378)的蔗糖中以产生溶液基质,用去离子水补至250ml。通过将0.065g来自aldrich(目录号435007)的羟丙基纤维素加入1ml去离子水中,制备6.5%的hpc(80000)溶液。如下制备用于微分配在前述凝固芯片上的aptt混合液:对于375μl的总体积,取75μl的aptt“试剂”,加入6.6μl的1mhepes、13.4μl的40%鼠李糖溶液、55μl的溶液基质、100μl的6.5%hpc(80000)、125μl的2mmaptt“底物”rf16-1。将该物质微分配到aptt芯片上并装配进筒装置中用于试验。图59显示了使用aptt测定试剂和全血得到的电流相对于时间的计时电流测定图(e.在含有甲基环糊精的去离子水中制备的鞣花酸)。在图59中显示的稳定化的鞣花酸溶液能够产生与基线鞣花酸制品方法(a.通过ph休克方法制备的仅鞣花酸溶液)类似的凝固曲线和时间。证实了稳定的鞣花酸的老化制品会产生可接受的凝固参数(数据未显示)。尽管已经以各种优选实施方案的方式描述了本发明,本领域技术人员会认识到,可以做出各种修改、置换、省略和变化,而不脱离本发明的精神。本发明的范围意图仅由下述权利要求的范围限制。另外,本领域技术人员应当理解,多个如上所述的本发明的不同实施方案可以彼此结合并整合进单个读出装置中。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1