一种测定三甲基镓中痕量氯离子的离子色谱方法与流程
本发明涉及测定痕量氯离子的离子色谱方法,尤其涉及测定三甲基镓中痕量氯离子的离子色谱方法,属于化学分析技术领域。
背景技术:
三甲基镓(TMG)是MO源材料中的重要的金属源,其运用到LED产业中能有效减少照明等一系列领域的耗电作用,有着巨大的经济效益。由于TMG的制备合成工艺中使用到卤代化合物—氯代化合物,残留的氯离子的含量会严重影响TMG的性能。为确保产品的性能,需要建立一种方法对TMG产品以及生产过程中的痕量氯离子进行测定。
关于TMG中痕量氯离子的报道未见相关报道,因此建立一种前处理方法简单、易重复,且方法灵敏度高,同时解决了测定过程中重金属镓对色谱柱及抑制器污染损坏的问题的测定TMG中痕量氯离子的离子色谱方法具有非常重要的意义。
技术实现要素:
本发明的目的是提供一种离子色谱测定TMG中痕量氯离子的前处理方法,该方法操作简单,重复性好,且灵敏度高。
本发明达到上述目的是通过以下技术方案实现的:
本发明首先公开了一种测定三甲基镓中痕量氯离子的离子色谱方法,包括以下步骤:
(1)将待测样品进行前处理:将待检测的三甲基镓样品溶液的pH值调为7.0-8.0后上前处理柱进行过柱处理;
(2)配置工作曲线溶液,进行离子色谱分析,得到不同浓度下Cl-的峰面积,以氯离子的浓度为横坐标,以其峰面积为纵坐标绘制工作曲线;
(3)将前处理后的三甲基镓样品溶液行离子色谱分析,得到Cl-的峰面积;
(4)根据工作曲线和Cl-的峰面积,得到三甲基镓中Cl-的含量。
本发明进一步提供了一种测定三甲基镓中痕量氯离子的离子色谱方法,包括以下步骤:
(1)将待测样品进行前处理:将待检测的三甲基镓样品溶液的pH值调为7.0-8.0后上前处理柱进行过柱处理;
(2)配置标准曲线溶液;
(3)将标准曲线溶液与前处理后的三甲基镓样品溶液在相同色谱条件下进行离子色谱分析,分别得到Cl-的峰面积,进行比较,即得。
其中,所述pH值为7.0。
由于样品经过5%稀硫酸溶解的溶液呈酸性;如果在该状态下进样发现由于游离的H+含量太高,导致了背景电导很高,从而使基线漂移较大,影响氯离子的定量;此外样品溶液中氯离子的保留时间比氯离子标准溶液的保留时间相延迟2min。为了保证样品溶液中氯离子测定结果的准确度,使得样品溶液中氯离子的保留时间和标准溶液中氯离子的保留时间的一致性,本发明对样品溶液的pH值进行了调节,最终结果表明当样品溶液的PH值调至接近中性(pH=7),氯离子的测定结果不受基线漂移的影响,且样品溶液中氯离子的保留时间与标准溶液中氯离子的保留时间最接近(相差0.09min)。
所述样品前处理柱的类型为H柱,其容量为2.5cc;
由于TMG样品中含有的大量的镓会对色谱柱和抑制器造成损害,因此进入离子色谱系统前,需要将样品溶液过H型前处理柱。消除重金属镓的原理:由于H型前处理柱中的填料为磺酸型阳离子交换树脂(即表面有大量的H+),H+和样品溶液中的镓离子进行置换反应,样品溶液中的镓离子被结合到H型前处理柱上,从而达到了去除样品溶液中重金属镓的目的。
所述离子色谱分析中所用的淋洗液为碳酸钠和碳酸氢钠混合溶液;优选的,所述的淋洗液为浓度1.8mmol/L的碳酸钠溶液和浓度为1.7-1.6mmol/L的碳酸氢钠溶液;更优选的,所述的淋洗液为浓度1.8mmol/L的碳酸钠和浓度为1.7mmol/L的碳酸氢钠溶液。
为确保氯离子测定准确度,必须保证氯离子色谱峰不受其他峰的干扰,即与相邻色谱峰之间的分离度达到1.5以上;该实验中,氯离子的干扰主要来源于样品溶液中的氢氧根离子,因此可通过调整淋洗液中碳酸氢根的浓度,改变氢氧根的保留时间。
在选定色谱柱的前提下,本发明分别对1.8/1.0,1.9/1.7,1.8/1.8,1.8/1.7,1.8/1.6和1.8/2.5(碳酸钠/碳酸氢钠)的色谱条件进行了试验,得到六种色谱条件下氯离子与相邻色谱峰之间的分离度及分析完成时间。试验结果表明,1.8/1.0和1.8/1.7(碳酸钠/碳酸氢钠)满足测定分离度的要求,而从整个过程分析时间来看,选择1.8/1.7的色谱条件,其分析效率比其它的条件有显著提升,因此最终选择的淋洗液为浓度1.8mmol/L的碳酸钠溶液和浓度为1.7mmol/L碳酸氢钠的溶液。
本发明技术方案与现有技术相比具有以下有益效果:
本发明离子色谱测定TMG中痕量氯离子的前处理方法操作简单,重复性好,且灵敏度高,能有效测定TMG中氯离子含量,同时解决了测定过程中重金属镓对色谱柱及抑制器污染损坏的问题。
附图说明
图1标准加入10μg/L氯离子溶液色谱图;
图2标准加入法工作曲线;
图3氯离子标准溶液色谱图;
图4样品溶液色谱图。
具体实施方式
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但是应理解所述实施例仅是范例性的,不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改或替换均落入本发明的保护范围。
1.1仪器和试剂
WY-IC6200型离子色谱仪;阴离子抑制器;电导检测器;H型前处理柱(青岛艾力析实验科技);
TMG样品(客户提供);碳酸钠(基准试剂);碳酸氢钠(优级纯);氢氧化钠(优级纯)。
1.2色谱条件
氯离子准储备溶液1000mg/L;流速:1.0mL/min;电导检测器温度:40℃;柱温:50℃;抑制电流:30mA;进样量:20μL。
1.3溶液的配置
(1)淋洗液的配制
精确称量0.1918g碳酸钠基准试剂和0.1428g碳酸氢钠,置于1000mL容量瓶中,用二次去离子水溶解并定容至刻度,得到1.8mmol/L的碳酸钠和1.7mmol/L碳酸氢钠的混合溶液,以此作为淋洗液。
(2)标准工作溶液的配制
精确移取50μL Cl-标准储备溶液50mL容量瓶中,用二次去离子水定容至刻度,得到Cl-浓度为1.0mg/L。
(3)空白溶液的配制
精确移取1.0mL5%的硫酸溶液,用0.125mol/L的氢氧化钠溶液稀释至10mL。
(4)标准加入法工作曲线溶液的配制
精确移取0、10、20、50、100和200μL 1.0μg/mL Cl-的标准溶液至6个2mL样品瓶中,并分别加入1000、990、980、950、900和800μL的空白溶液,混匀,得到标准加入法的Cl-的浓度分别为0、10、20、50、100和200μg/L。
(5)标准曲线溶液的配制
精确移取100μL 1.0μg/mL氯离子的标准工作溶液,加入900μL二次去离子水,混匀,得到其浓度为100μg/L。
(6)重复性溶液的配制
精确移取10.0μL标准工作溶液,加入990μL空白溶液,混匀,即得加标10μg/L的Cl-溶液。
实验例1淋洗液的碳酸钠/碳酸氢钠比例的确定
为确保氯离子测定准确度,必须保证氯离子色谱峰不受其他峰的干扰,即与相邻色谱峰之间的分离度达到1.5以上;该实验中,氯离子的干扰主要来源于样品溶液中的氢氧根离子,因此可通过调整淋洗液中碳酸氢根的浓度,改变氢氧根的保留时间。
在选定色谱柱的前提下,本方法分别对1.8/1.0,1.9/1.7,1.8/1.8,1.8/1.7,1.8/1.6和1.8/2.5(碳酸钠/碳酸氢钠)的色谱条件进行了试验,得到六种色谱条件下氯离子与相邻色谱峰之间的分离度及分析完成时间,见表1。
从表1中可以看出1.8/1.0,1.8/1.8,1.8/1.6和1.8/1.7(碳酸钠/碳酸氢钠)基本满足测定分离度的要求,而从整个过程分析时间来看,1.8/1.7和1.8/1.8分析时间较短,结合分离度数据,最后选择1.8/1.7的色谱条件,将氯离子受相邻色谱峰干扰的影响降到很低,同时其分析效率将提高很多,因此最终选择的淋洗液浓度为1.8mmol/L的碳酸钠的溶液和浓度为1.7mmol/L的碳酸氢钠的溶液。
表1淋洗液对色谱分析的影响
实验例2氢氧化钠体积的确定
样品经过5%稀硫酸溶解的溶液呈酸性;如果在该状态下进样发现由于游离的H+含量太高,导致了背景电导很高,从而使基线漂移较大,影响氯离子的定量;此外样品溶液中氯离子的保留时间比氯离子标准溶液的保留时间相延迟2min。为了保证样品溶液中氯离子测定结果的准确度,及样品溶液中氯离子的保留时间和标准溶液中氯离子的保留时间的一致性,利用氢氧化钠溶液调节样品溶液的pH值。
取4份相同的样品溶液分别以1:6,1:8,1:10,1:12(样品溶液:氢氧化钠溶液,V/V)的稀释比进行实验不同体积比的样品溶液中氯离子保留时间与标准溶液保留时间的差值,见表2。
通过实验发现1:10稀释比(即氢氧化钠溶液的加入量为10mL),调至接近中性(pH=7)时,氯离子的测定结果不受基线漂移的影响,且样品溶液中氯离子的保留时间与标准溶液中氯离子的保留时间最接近(相差0.09min)。
表2氢氧化钠溶液加入量对色谱分析的影响
实验例3TMG中痕量氯离子测定
1、样品溶液的前处理
(1)在避光条件下,精确称取TMG样品的质量为0.2000g,置于100mL容量瓶中;
(2)用滴定管迅速加入5%的硫酸溶液将其溶解,并用5%的硫酸溶液定容至刻度,得到TMG的样品溶液;
(3)精确移取1.0mL(2)中的样品溶液,置于10mL容量瓶中,用0.125mol/L的氢氧化钠溶液稀释并定容至刻度,得到TMG的供试品溶液;
(4)取(3)中的供试品溶液过样品H型前处理柱后进入离子色谱系统进行分析,其中样品的前3mL样品溶液要弃去。
其中由于TMG样品中含有的大量的镓会对色谱柱和抑制器造成损害,因此进入离子色谱系统前,需要将样品溶液过H型前处理柱,即:H型前处理柱使用前须使用15mL二次去离子水进行活化,活化方法为:采用5mL注射器以2.0mL/min的速度进行冲洗,冲洗的体积为15mL,并对最后冲洗出来的二次去离子水进行测定,若测定含有Cl-,则需要继续进行冲洗,直到冲洗出来的二次去离子水中Cl-不被检出。
消除重金属镓的原理:由于H型前处理柱中的填料为磺酸型阳离子交换树脂(即表面有大量的H+),H+和样品溶液中的镓离子进行置换反应,样品溶液中的镓离子被结合到H型前处理柱上,从而达到了去除样品溶液中重金属镓的目的。
2、标准加入法溶液的分析
(1)重复性
对重复性溶液连续进样6次,得到Cl-标准加入溶液分析的色谱图(见图1),并得到氯离子色谱峰的保留时间和峰面积的相对标准偏差分别为0.27%和0.70%。
(2)线性关系
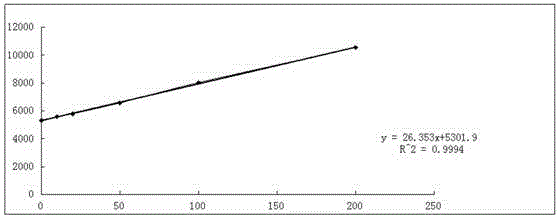
对配制的标准加入法的线性溶液,依据从低浓度到高浓度的进样顺序进行进样分析,得到不同浓度下Cl-的峰面积,以氯离子的浓度(mg/L)为横坐标,以其峰面积为纵坐标绘制工作曲线,见图2。
3、标准曲线溶液的分析
对配制的100μg/L的Cl-标准溶液进样分析,得到Cl-的色谱图,见图3。
4、TMG样品中氯离子含量的测定及加标回收率
在选定的色谱条件下,对处理好的TMG样品溶液进行检测,得到实际样品检测的色谱图,见图4,同时分别根据标准加入法和单点外标法对实际样品中的Cl-进行定量,并得到采用两种计算方法的样品溶液中Cl-浓度,测得结果见表3,从表中可以看出,根据标准加入法和标准曲线法计算得到氯离子的浓度的相对标准偏差在1.2~7.99%之间,证明了这两种方法在针对TMG中氯离子的测定中结果一致性理想。
对实际样品进行加标回收率试验,分别测得加标10.0、20.0及100μg/L的样品回收率,测得结果见表4,从表中可以得到回收率在84.2~106.6%之间,验证了该实验方法的可靠性。
表3样品溶液中Cl-浓度的测定结果
表4对样品进行加标回收率试验样品回收率测定结果
- 还没有人留言评论。精彩留言会获得点赞!