利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法及应用与流程
本发明属于药物检测领域,具体涉及利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法及应用。
背景技术:
气滞胃痛颗粒由柴胡、延胡索、枳壳、香附、白芍和炙甘草6味中药制成,具有舒肝理气、和胃止痛的作用,用于肝郁气滞、胸痞胀满、胃脘疼痛等疾病的治疗,经过多年临床实践,现已成为临床治疗胃脘痛的一线用药。但是,气滞胃痛颗粒的制备过程复杂而繁琐,实际生产过程中各种工艺参数的微小变化都有可能直接影响最终药物的质量。
传统的药物分析技术一般采用离线的分析手段,从提取罐和浓缩罐中采集药液,在实验室中通过高效液相色谱、气相色谱等进行分析,然后将分析后的结果反馈回生产车间进行生产调控。然而,传统分析方式由于效率低下,不仅大大影响了生产效率,而且很难反映出提取过程和浓缩过程中复杂的化学变化和物理变化。在推进中药产业现代化发展的进程中,随着我国药品生产过程自动化程度的不断提高,为了保证中药产品质量的均一性和稳定性,研究中药制药过程的快速质量分析方法与在线检测技术,对于提高我国中药产业现代化水平具有重要意义。
近红外光谱(nearinfraredspectruminstrument,nirs)是近年迅速发展起来的一种有效简便的分析方法,通过与光导纤维和计算机技术相结合,nir测量信号可以进行远距离传输和分析。近红外光是一种电磁波,介于可见光(vis)和中红外(mir)之间的电磁辐射波,波长在780-2526nm,近红外光谱分析具有不污染环境、无损取样、分析速度快、操作简单、实时在线性、综合评价、准确度高、分析对象广泛、无相关耗材和维修成本等特点。近红外光谱区与有机分子中含氢基团(o-h、n-h、c-h)振动的合频和各级倍频的吸收区一致,通过扫描样品的近红外光谱,可以得到样品中有机分子含氢基团的特征信息,利用近红外光谱技术分析样品的最大特点是快速、无损、原位在线等,因此,该技术越来越受到研究人员的青睐。目前,近红外光谱技术已在医药、石油、食品工业等领域获得较成功的应用。
因此,建立一种能够快速地、全面地检测气滞胃痛颗粒的制备过程的方法,对气滞胃痛颗粒的大批量生产和全面质量控制具有重要意义。
技术实现要素:
为此,本发明提供一种利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法及应用。
为解决上述技术问题,本发明是通过以下技术方案来实现的:
本发明提供一种利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,
包括如下气滞胃痛颗粒的水提取过程的检测和/或气滞胃痛颗粒的提取挥发油过程的检测和/或气滞胃痛颗粒的提取液浓缩过程的检测:
a、气滞胃痛颗粒的水提取过程的检测
包括如下芍药苷的含量测定和/或固含量测定的步骤:
a、芍药苷的含量测定包括以下步骤:
(1)取已知芍药苷含量的气滞胃痛颗粒的水提取液,备用;
(2)将所述气滞胃痛颗粒的水提取液进行近红外光谱扫描,采集所述气滞胃痛颗粒的水提取液的近红外光谱;
(3)选取5448.8~6102.7cm-1和6201~6501cm-1特征波段下的光谱信息,应用化学计量学软件与所述已知芍药苷含量的气滞胃痛颗粒的水提取液的芍药苷含量进行关联,采用偏最小二乘法建立近红外光谱与标准含量之间的定量校准模型;
(4)按照所述步骤(2)的方法对未知气滞胃痛颗粒的水提取液样品进行近红外光谱扫描,并选取5448.8~6102.7cm-1和6201~6501cm-1特征波段下的光谱信息,导入建立的定量校准模型获得所述未知气滞胃痛颗粒的水提取液样品的芍药苷含量值;
b、固含量测定包括以下步骤:
(1)取已知固含量的气滞胃痛颗粒的水提取液,备用;
(2)将所述气滞胃痛颗粒的水提取液进行近红外光谱扫描,采集所述气滞胃痛颗粒的水提取液的近红外光谱;
(3)选取6201~5701cm-1和7170~7300cm-1特征波段下的光谱信息,应用化学计量学软件与所述已知固含量的气滞胃痛颗粒的水提取液的固含量进行关联,采用偏最小二乘法建立近红外光谱与标准含量之间的定量校准模型;
(4)按照所述步骤(2)的方法对未知气滞胃痛颗粒的水提取液样品进行近红外光谱扫描,并选取6201~5701cm-1和7170~7300cm-1特征波段下的光谱信息,导入建立的定量校准模型获得所述未知气滞胃痛颗粒的水提取液样品的固含量值;
b、气滞胃痛颗粒的提取挥发油过程的检测
包括如下柚皮苷的含量测定、新橙皮苷的含量测定和/或固含量测定的步骤:
a、柚皮苷的含量测定包括以下步骤:
(1)取已知柚皮苷含量的气滞胃痛颗粒的提取挥发油过程的提取液,备用;
(2)将所述气滞胃痛颗粒的提取挥发油过程的提取液进行近红外光谱扫描,采集所述气滞胃痛颗粒的提取挥发油过程的提取液的近红外光谱;
(3)选取5500~6100cm-1特征波段下的光谱信息,应用化学计量学软件与所述已知柚皮苷含量的气滞胃痛颗粒的提取挥发油过程的提取液的柚皮苷含量进行关联,采用偏最小二乘法建立近红外光谱与标准含量之间的定量校准模型;
(4)按照所述步骤(2)的方法对未知气滞胃痛颗粒的提取挥发油过程的提取液样品进行近红外光谱扫描,并选取5500~6100cm-1特征波段下的光谱信息,导入建立的定量校准模型获得所述未知气滞胃痛颗粒的提取挥发油过程的提取液样品的柚皮苷含量值;
b、新橙皮苷的含量测定包括以下步骤:
(1)取已知新橙皮苷含量的气滞胃痛颗粒的提取挥发油过程的提取液,备用;
(2)将所述气滞胃痛颗粒的提取挥发油过程的提取液进行近红外光谱扫描,采集所述气滞胃痛颗粒的提取挥发油过程的提取液的近红外光谱;
(3)选取7170~7270cm-1和5500~6100cm-1特征波段下的光谱信息,应用化学计量学软件与所述已知新橙皮苷含量的气滞胃痛颗粒的提取挥发油过程的提取液的新橙皮苷含量进行关联,采用偏最小二乘法建立近红外光谱与标准含量之间的定量校准模型;
(4)按照所述步骤(2)的方法对未知气滞胃痛颗粒的提取挥发油过程的提取液样品进行近红外光谱扫描,并选取7170~7270cm-1和5500~6100cm-1特征波段下的光谱信息,导入建立的定量校准模型获得所述未知气滞胃痛颗粒的提取挥发油过程的提取液样品的新橙皮苷含量值;
c、固含量测定包括以下步骤:
(1)取已知固含量的气滞胃痛颗粒的提取挥发油过程的提取液,备用;
(2)将所述气滞胃痛颗粒的提取挥发油过程的提取液进行近红外光谱扫描,采集所述气滞胃痛颗粒的提取挥发油过程的提取液的近红外光谱;
(3)选取5600~6000cm-1特征波段下的光谱信息,应用化学计量学软件与所述已知固含量的气滞胃痛颗粒的提取挥发油过程的提取液的固含量进行关联,采用偏最小二乘法建立近红外光谱与标准含量之间的定量校准模型;
(4)按照所述步骤(2)的方法对未知气滞胃痛颗粒的提取挥发油过程的提取液样品进行近红外光谱扫描,并选取5600~6000cm-1特征波段下的光谱信息,导入建立的定量校准模型获得所述未知气滞胃痛颗粒的提取挥发油过程的提取液样品的固含量值;
c、气滞胃痛颗粒的提取液浓缩过程的检测
包括如下芍药苷的含量测定、相对密度和/或固含量测定的步骤:
a、芍药苷的含量测定包括以下步骤:
(1)取已知芍药苷含量的气滞胃痛颗粒的浓缩提取液,备用;
(2)将所述气滞胃痛颗粒的浓缩提取液进行近红外光谱扫描,采集所述气滞胃痛颗粒的浓缩提取液的近红外光谱;
(3)选取5449.8~6101.7cm-1和5149.01~5442.13cm-1特征波段下的光谱信息,应用化学计量学软件与所述已知芍药苷含量的气滞胃痛颗粒的浓缩提取液的芍药苷含量进行关联,采用偏最小二乘法建立近红外光谱与标准含量之间的定量校准模型;
(4)按照所述步骤(2)的方法对未知气滞胃痛颗粒的浓缩提取液样品进行近红外光谱扫描,并选取5449.8~6101.7cm-1和5149.01~5442.13cm-1特征波段下的光谱信息,导入建立的定量校准模型获得所述未知气滞胃痛颗粒的浓缩提取液样品的芍药苷含量值;
b、相对密度测定包括以下步骤:
(1)取已知相对密度的气滞胃痛颗粒的浓缩提取液,备用;
(2)将所述气滞胃痛颗粒的浓缩提取液进行近红外光谱扫描,采集所述气滞胃痛颗粒的浓缩提取液的近红外光谱;
(3)选取5469.13~7143.04cm-1和5149.01~5442.13cm-1特征波段下的光谱信息,应用化学计量学软件与所述已知相对密度的气滞胃痛颗粒的浓缩提取液的相对密度进行关联,采用偏最小二乘法建立近红外光谱与标准含量之间的定量校准模型;
(4)按照所述步骤(2)的方法对未知气滞胃痛颗粒的浓缩提取液样品进行近红外光谱扫描,并选取5469.13~7143.04cm-1和5149.01~5442.13cm-1特征波段下的光谱信息,导入建立的定量校准模型获得所述未知气滞胃痛颗粒的浓缩提取液样品的相对密度值;
c、固含量测定包括以下步骤:
(1)取已知固含量的气滞胃痛颗粒的浓缩提取液,备用;
(2)将所述气滞胃痛颗粒的浓缩提取液进行近红外光谱扫描,采集所述气滞胃痛颗粒的浓缩提取液的近红外光谱;
(3)选取5469.13~7143.04cm-1特征波段下的光谱信息,应用化学计量学软件与所述已知固含量的气滞胃痛颗粒的浓缩提取液的固含量进行关联,采用偏最小二乘法建立近红外光谱与标准含量之间的定量校准模型;
(4)按照所述步骤(2)的方法对未知气滞胃痛颗粒的浓缩提取液样品进行近红外光谱扫描,并选取5469.13~7143.04cm-1特征波段下的光谱信息,导入建立的定量校准模型获得所述未知气滞胃痛颗粒的浓缩提取液样品的固含量值。
优选地,本发明上述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,
所述气滞胃痛颗粒的水提取过程的检测中的芍药苷的含量测定的步骤(2)中和/或固含量测定的步骤(2)中,采用静态光谱采集法进行所述气滞胃痛颗粒的水提取液的近红外光谱采集,具体条件为:光谱采集时间间隔为10min,以空气为参比,扫描次数为32次,分辨率为8cm-1,扫描光谱范围为4000~10000cm-1;
所述气滞胃痛颗粒的提取挥发油过程的检测中的柚皮苷的含量测定的步骤(2)中、新橙皮苷的含量测定的步骤(2)中和/或固含量测定的步骤(2)中,采用静态光谱采集法进行所述气滞胃痛颗粒的提取挥发油过程的近红外光谱采集,具体条件为:光谱采集时间间隔为10min,以空气为参比,扫描次数为32次,分辨率为8cm-1,扫描光谱范围为4000~10000cm-1;
所述气滞胃痛颗粒的提取液浓缩过程的检测中的芍药苷的含量测定的步骤(2)中和/或固含量测定的步骤(2)中,采用静态光谱采集法进行所述气滞胃痛颗粒的浓缩提取液的近红外光谱采集,具体条件为:光谱采集时间间隔为10min,以空气为参比,扫描次数为32次,分辨率为8cm-1,扫描光谱范围为4000~10000cm-1。
进一步优选地,本发明上述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,
所述气滞胃痛颗粒的水提取过程的检测中的芍药苷的含量测定的步骤(2)中和/或固含量测定的步骤(2)中,还包括对采集到的所述近红外光谱采用一阶导数法和norris导数平滑滤波法进行预处理的步骤;
所述气滞胃痛颗粒的提取挥发油过程的检测中的柚皮苷的含量测定的步骤(2)中和/或新橙皮苷的含量测定的步骤(2)中,还包括对采集到的所述近红外光谱采用一阶导数法和norris导数平滑滤波法进行预处理的步骤;
所述气滞胃痛颗粒的提取挥发油过程的检测中的固含量测定的步骤(2)中,还包括对采集到的所述近红外光谱采用一阶导数法和s-g平滑滤波法进行预处理的步骤;
所述气滞胃痛颗粒的提取液浓缩过程的检测中的芍药苷的含量测定的步骤(2)中,还包括对采集到的所述近红外光谱采用二阶导数法和norris导数平滑滤波法进行预处理的步骤;
所述气滞胃痛颗粒的提取液浓缩过程的检测中的相对密度的步骤(2)中和/或固含量测定的步骤(2)中,还包括对采集到的所述近红外光谱采用一阶导数法和norris导数平滑滤波法进行预处理的步骤。
本发明气滞胃痛颗粒的原料药组成和制备方法参照中国专利文献cn1712048a。
进一步优选地,本发明上述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,
所述气滞胃痛颗粒的原料药组成为:
柴胡30~100重量份、甘草15~60重量份、香附35~120重量份、延胡索35~120重量份、白芍40~150重量份、枳壳35~120重量份。
进一步优选地,本发明上述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,
所述气滞胃痛颗粒的制备方法包括以下步骤:
(3)取选定重量份的枳壳和香附,提取挥发油,药液备用,药渣弃去;
(4)取选定重量份的柴胡、甘草、延胡索和白芍,加水煎煮2次,第1次煎煮0.5~4小时,第2次煎煮0.5~3小时;
(3)合并步骤(1)和步骤(2)的煎液,过滤,滤液浓缩至相对密度为1.10~1.30的清膏;
(4)取清膏,加辅料,混匀制成颗粒,干燥,喷入挥发油,即得颗粒剂。
进一步优选地,本发明上述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,
所述气滞胃痛颗粒的水提取过程的检测中的芍药苷的含量测定的步骤(3)中和/或固含量测定的步骤(3)中,还包括对建立的所述定量校准模型的预测性能进行评价的步骤,所述评价指标包括相关系数r、校正集均方差rmsec、验证集均方根rmsep、交叉验证均方根rmsecv和预测相对偏差rsep,当r值越接近于1,rmsec和rmsep值越小且越接近时,评价模型稳定性越佳、预测精准度越高,则能够满足气滞胃痛颗粒直接分析的预测精度要求;反之,则不适用;
所述气滞胃痛颗粒的提取挥发油过程的检测中的柚皮苷的含量测定的步骤(3)中、新橙皮苷的含量测定的步骤(3)中和/或固含量测定的步骤(3)中,还包括对建立的所述定量校准模型的预测性能进行评价的步骤,所述评价指标包括相关系数r、校正集均方差rmsec、验证集均方根rmsep、交叉验证均方根rmsecv和预测相对偏差rsep,当r值越接近于1,rmsec和rmsep值越小且越接近时,评价模型稳定性越佳、预测精准度越高,则能够满足气滞胃痛颗粒直接分析的预测精度要求;反之,则不适用;
所述气滞胃痛颗粒的提取液浓缩过程的检测中的芍药苷的含量测定的步骤(3)中、相对密度测定的步骤(3)中和/或固含量测定的步骤(3)中,还包括对建立的所述定量校准模型的预测性能进行评价的步骤,所述评价指标包括相关系数r、校正集均方差rmsec、验证集均方根rmsep、交叉验证均方根rmsecv和预测相对偏差rsep,当r值越接近于1,rmsec和rmsep值越小且越接近时,评价模型稳定性越佳、预测精准度越高,则能够满足气滞胃痛颗粒直接分析的预测精度要求;反之,则不适用。
进一步优选地,本发明上述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,
所述气滞胃痛颗粒的水提取过程的检测中的芍药苷的含量测定的步骤(1)中和/或所述气滞胃痛颗粒的提取液浓缩过程的检测中的芍药苷的含量测定的步骤(1)中,采用高效液相色谱法测定所述已知芍药苷含量气滞胃痛颗粒的芍药苷含量作为标准含量;
所述气滞胃痛颗粒的水提取过程的检测中的固含量测定的步骤(3)中、所述气滞胃痛颗粒的提取挥发油过程的检测中的固含量测定的步骤(3)中和/或所述气滞胃痛颗粒的提取液浓缩过程的检测中的固含量测定的步骤(3)中,采用烘干法测定所述已知固含量气滞胃痛颗粒的固含量作为标准含量;
所述气滞胃痛颗粒的提取挥发油过程的检测中的柚皮苷的含量测定的步骤(3)中,采用高效液相色谱法测定所述已知柚皮苷含量气滞胃痛颗粒的柚皮苷含量作为标准含量;
所述气滞胃痛颗粒的提取挥发油过程的检测中的新橙皮苷的含量测定的步骤(3)中,采用高效液相色谱法测定所述已知新橙皮苷含量气滞胃痛颗粒的新橙皮苷含量作为标准含量。
进一步优选地,本发明上述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,
所述采用高效液相色谱法测定所述已知芍药苷含量气滞胃痛颗粒的芍药苷含量的具体步骤为:
(1)取所述气滞胃痛颗粒的水提取液4~6ml或所述气滞胃痛颗粒的浓缩提取液4~6ml,在转速12000-14000r/min条件下离心8~12min,取上清液,作为供试品溶液;
(2)精密称取1-3mg芍药苷对照品置于20ml量瓶中,加甲醇制成每ml含0.05~0.15mg的溶液,摇匀,作为对照品溶液;
(3)色谱条件:以4.6×250mm、5μm的agilentzorbaxsb-c18为色谱柱,以0.1%磷酸水溶液为流动相a、以乙腈为流动相b按照如下程序进行梯度洗脱:0-13min,a:b为85%:15%;13-15min,a:b为85%:15%→10%:90%;15-19min,a:b为10%:90%;19-25min,a:b为10%:90%→0%:100%;25-30min,a:b为0%:100%→85%:15%;流速1.0ml/min,柱温30℃,检测波长230nm;
(4)精密吸取所述供试品溶液和对照品溶液各5μl,注入液相色谱仪,测定;
采用高效液相色谱法测定所述已知柚皮苷含量气滞胃痛颗粒的柚皮苷含量的具体步骤为:
(1)取所述所述气滞胃痛颗粒的提取挥发油过程的提取液4~6ml,在转速12000-14000r/min条件下离心8~12min,取上清液,作为供试品溶液;
(2)精密称取1-3mg柚皮苷对照品置于20ml量瓶中,加甲醇制成每ml含0.05~0.15mg的溶液,摇匀,作为对照品溶液;
(3)色谱条件:以4.6×250mm、5μm的agilentzorbaxsb-c18为色谱柱,以0.1%磷酸水溶液为流动相a、以乙腈为流动相b按照如下程序进行梯度洗脱:0-18min,a:b为80%:20%;18-21min,a:b为80%:20%→10%:90%;21-25min,a:b为10%:90%;21-30min,a:b为10%:90%→0%:100%;30-35min,a:b为0%:100%→80%:20%;流速1.0ml/min,柱温30℃,检测波长283nm;
(4)精密吸取所述供试品溶液和对照品溶液各5μl,注入液相色谱仪,测定;
采用高效液相色谱法测定所述已知新橙皮苷含量气滞胃痛颗粒的新橙皮苷含量的具体步骤为:
(1)取所述所述气滞胃痛颗粒的提取挥发油过程的提取液4~6ml,在转速12000-14000r/min条件下离心8~12min,取上清液,作为供试品溶液;
(2)精密称取1-3mg新橙皮苷对照品置于20ml量瓶中,加甲醇制成每ml含0.05~0.15mg的溶液,摇匀,作为对照品溶液;
(3)色谱条件:以4.6×250mm、5μm的agilentzorbaxsb-c18为色谱柱,以0.1%磷酸水溶液为流动相a、以乙腈为流动相b按照如下程序进行梯度洗脱:0-18min,a:b为80%:20%;18-21min,a:b为80%:20%→10%:90%;21-25min,a:b为10%:90%;21-30min,a:b为10%:90%→0%:100%;30-35min,a:b为0%:100%→80%:20%;流速1.0ml/min,柱温30℃,检测波长283nm;
(4)精密吸取所述供试品溶液和对照品溶液各5μl,注入液相色谱仪,测定;
所述采用烘干法测定所述已知固含量气滞胃痛颗粒的固含量的具体步骤为:
取4~6ml所述气滞胃痛颗粒的水提取液或4~6ml所述气滞胃痛颗粒的提取挥发油过程的提取液或4~6ml所述气滞胃痛颗粒的浓缩提取液至于烘干至恒重的称量瓶,于105℃烘干5~7h,移至干燥器中冷却20~40min,称重;再于105℃下干燥0.5~1.5小时,冷却,称重;至连续两次称重差不超过5mg为止,计算固含量。
本发明还提供上述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法在气滞胃痛颗粒质量检测及控制中的用途。
与现有技术相比,本发明的上述技术方案具有以下优点:
(1)本发明所述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,将近红外光谱法应用于气滞胃痛颗粒的制备过程的质量快速检测中,针对水提取过程、提取挥发油过程和提取液浓缩过程分别确定质控指标,能够对气滞胃痛颗粒的各制备过程(水提取过程、提取挥发油过程和提取液浓缩过程)快速测定,实现了对气滞胃痛颗粒的制备过程快速的、全面的检测,并且具有操作简单、准确性高、精确度高等优点,能快速判断气滞胃痛颗粒的制备过程(水提取过程、提取挥发油过程和提取液浓缩过程)是否合格,缩短检测时间,节约生产成本,提高生产效率和经济效益,保证了气滞胃痛颗粒质量的安全、有效;
(2)本发明所述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,通过选择气滞胃痛颗粒的制备过程(水提取过程、提取挥发油过程和提取液浓缩过程)中的各质控指标的近红外光谱中的光谱波段,提取有效的特征光谱波段,该特征光谱波段与按照现有常规方法测定的各质控指标具有良好的相关性,可有效监控气滞胃痛颗粒制备过程(水提取过程、提取挥发油过程和提取液浓缩过程)是否合格;
(3)本发明所述利用近红外光谱法快速检测气滞胃痛颗粒的制备过程的方法,针对制备过程(水提取过程、提取挥发油过程和提取液浓缩过程)中的各质控指标分别优化了近红外原始光谱的预处理方法,以筛选信息,减少噪音,提高了该方法检测气滞胃痛颗粒的制备过程的准确性和精确度;
(4)本发明建立了气滞胃痛颗粒的制备过程中水提取过程、提取挥发油过程和提取液浓缩过程的近红外光谱在线分析方法,并将其应用于在线检测,实现了气滞胃痛颗粒的制备过程的自动化检测和各质控指标含量的实时预测;所建立的模型预测精度高,准确度高,满足实际生产中定量分析的要求;该方法与传统药典方法相比,在保证准确度的基础上,大大提高了分析效率,可以快速、高效地对提取过程的质量进行实时监测和控制,从而有利于保证最终产品质量的安全性、稳定性和有效性。
附图说明
为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中:
图1是实施例1中的气滞胃痛颗粒的水提取过程的原始近红外光谱图;
图2是实施例1中的芍药苷对照品的hplc色谱图;
图3是实施例1中的芍药苷样品的hplc色谱图;
图4是实施例1中的芍药苷校正集样品的近红外预测值与高效液相测定值的相关关系图;
图5是实施例1中的固含量校正集样品的近红外预测值与测定值的相关关系图;
图6是实施例1中的芍药苷验证集样品的近红外预测值与高效液相测定值的相关关系图;
图7是实施例1中的固含量验证集样品的近红外预测值与测定值的相关关系图;
图8是实施例1中的水提取过程一煎液相色谱测定与模型预测芍药苷的浓度变化对照图;
图9是实施例1中的水提取过程一煎固含量测定与模型预测固含量变化对照图;
图10是实施例1中的气滞胃痛颗粒的水提取过程二煎液相色谱测定与模型预测芍药苷的浓度变化对照图;
图11是实施例1中的气滞胃痛颗粒的水提取过程二煎固含量测定与模型预测固含量变化对照图;
图12是实施例2中的气滞胃痛颗粒的提取挥发油过程的原始近红外光谱;
图13是实施例2中的柚皮苷校正集样品的近红外预测值与高效液相测定值的相关关系图;
图14是实施例2中的新橙皮苷校正集样品的近红外预测值与高效液相测定值的相关关系图;
图15是实施例2中的固含量校正集样品的近红外预测值与测定值的相关关系图;
图16是实施例2中的柚皮苷验证集样品的近红外预测值与hplc测定值的相关关系图;
图17是实施例2中的新橙皮苷验证集样品的近红外预测值与hplc测定值的相关关系图;
图18是实施例2中的固含量验证集样品的近红外预测值与测定值的相关关系图;
图19是实施例2中的提取挥发油过程的液相色谱测定与模型预测柚皮苷的浓度变化对照图;
图20是实施例2中的提取挥发油过程的液相色谱测定与模型预测新橙皮苷的浓度变化对照图;
图21是实施例2中的提取挥发油过程的固含量测定与模型预测固含量变化对照图;
图22是实施例3中的气滞胃痛颗粒的提取液浓缩过程的原始近红外光谱;
图23是实施例3中的芍药苷对照品色谱图;
图24是实施例3中的芍药苷样品色谱图;
图25是实施例3中的芍药苷校正集样品的近红外预测值与高效液相测定值的相关关系图;
图26是实施例3中的相对密度校正集样品的近红外预测值与测定值的相关关系图;
图27是实施例3中的固含量校正集样品的近红外预测值与测定值的相关关系图;
图28是实施例3中的芍药苷验证集样品的近红外预测值与高效液相测定值的相关关系图;
图29是实施例3中的相对密度验证集样品的近红外预测值与测定值的相关关系图;
图30是实施例3中的固含量验证集样品的近红外预测值与测定值的相关关系图;
图31是实施例3中的提取液浓缩过程液相色谱测定与模型预测芍药苷的浓度变化对照图;
图32是实施例3中的提取液浓缩过程相对密度测定与模型预测相对密度的浓度变化对照图;
图33是实施例3中的提取液浓缩过程固含量测定与模型预测固含量的浓度变化对照图。
具体实施方式
实施例1近红外光谱技术用于气滞胃痛颗粒制备过程中水提取过程的研究
1.仪器与试药
1.1主要药材与试剂
柴胡、白芍、炙甘草、延胡索(炙)(辽宁华润本溪三药有限公司提供);芍药苷对照品(成都曼斯特生物科技有限公司,批号:must-11030608);乙腈(merck,germany);磷酸(aladdin);哇哈哈纯净水。
1.2主要仪器与设备
agilent1200高效液相色谱仪(agilent1200泵、agilent1200光电二级阵列检测器、agilent1200色谱工作站,美国安捷伦科技公司);antaris傅立叶变换近红外光谱仪(thermonicolet,usa);配有相应透射检测器附件采样系统和信号采集及result、tqanalyst等数据处理软件;sartoriuscp225d电子分析天平(北京赛多利斯天平有限公司);re-52aa旋转蒸发仪(上海亚荣生化仪器厂);40l多功能提取罐(浙江温兄机械阀业有限公司);hs6150型超声波清洗器(天津恒奥科技发展有限公司)。
2.方法与结果
2.1水提取液样品的采集
将包含白芍、柴胡、延胡索和甘草的4kg药材投入容积为40l的多功能提取罐中,按照生产工艺进行一煎、二煎提取。为了保证所采集的样品浓度均匀分布,一煎过程中,浸泡阶段每10min取样一次,加热阶段中每8min取样一次,沸腾过程中前30min每6min取样一次其余时间每10min取样一次;二煎过程中加热阶段每8min取样一次,沸腾过程每10min取样一次,共进行7个批次水提实验。所有批次实验完成后共得到210个样本。将第1、2、3、5、6、7次实验的提取液样品作为校正集,第4次提取样品作为验证集。
2.2近红外光谱采集
采集气滞胃痛颗粒的水提取过程的提取液样品的透射光谱,设定光谱的扫描范围4000~10000cm-1,扫描次数为32次,分辨率为8cm-1,以空气为背景,采用5mm光程的圆柱形玻璃比色管,在一个恒定的环境中(温度为25℃,湿度为35%-45%)采集气滞胃痛颗粒的水提取过程的提取液透射光谱,每个样品重复测定3次,计算平均光谱。气滞胃痛颗粒的水提取过程的的原始近红外光谱图如图1所示。
通过opus软件的自动优化功能,筛选出5448.8~6102.7cm-1和7498.9-8451.5cm-1两个波段,同时根据光谱变化趋势选取6201~6501cm-1之间的波段进行模型建立,通过优化比较,最后选择5448.8~6102.7cm-1和6201-6501cm-1两个波段建立水提取过程芍药苷定量检测模型。根据tq软件给出的建模波段,对于固含量模型选择6201~5701cm-1和7170~7300cm-1两个波段进行定量模型建立。并采用一阶、二阶导数处理、平滑、矢量归一、散射效应校正、多元线性校正、小波变换等多种化学计量学方法,本次模型建立采用一阶、二阶导数处理结合s-g或者norris平滑滤波的方法对光谱进行预处理。
表1和表2分别为采用原始光谱和不同预处理方法后建立芍药苷及固含量的近红外定量校正模型得到的r2、rmsec、rmsecv值,通过比较不同预处理方法所建立的模型的rmsec和rmsecv值确定预处理方法。
表1不同预处理方法的水提取过程芍药苷定量测定模型的参数
表2不同预处理方法的水提取过程固含量定量测定模型的参数
由表1可知,水提取过程芍药苷定量测定模型的预处理方法采用一阶导数处理+norris平滑滤波的方法时,rmsecv值和rmsec值最小;由表2可知,水提取过程固含量定量测定模型的预处理方法采用一阶导数处理+norris平滑滤波的方法时,rmsecv值和rmsec值最小。定量模型采用偏最小二乘法(plsr)进行计算。
2.3芍药苷含量测定
将上述采集到的提取液样品离心10min(13000r/min),移取上清液,进行hplc含量测定。hplc色谱条件如下:色谱柱:agilentzorbaxsb-c18(4.6×250mm,5μm);流动相:0.1%磷酸水(a)-乙腈(b);进样量:5μl;流速:1.0ml/min;柱温:30℃;检测波长:230nm。洗脱梯度程序如表3所示。
表3梯度洗脱程序
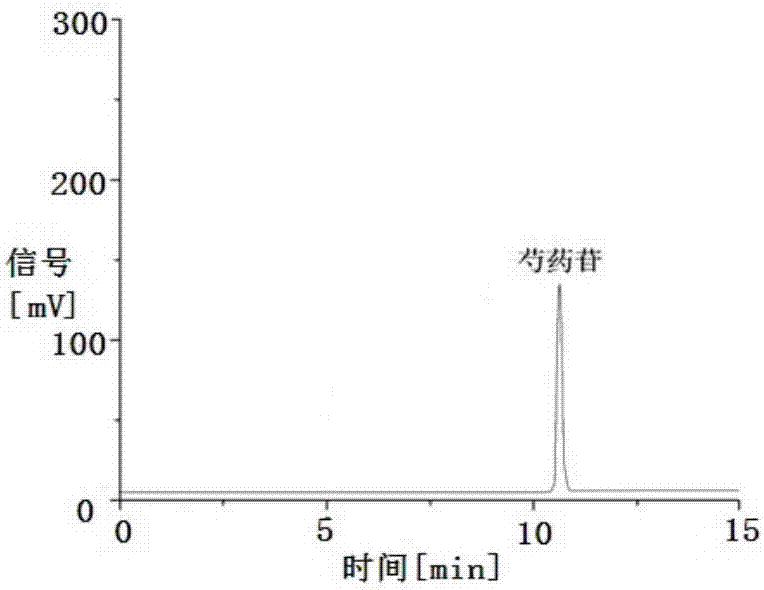
按2.3色谱条件分析,进样量为5μl,测定不同批次芍药苷的含量,结果见表4所示。芍药苷对照品的hplc色谱图如图2所示,芍药苷样品的hplc色谱图如图3所示。
表4水提取过程芍药苷含量测定结果
2.4固含量测定
提取液静置24h,量取4ml上清液至已烘干至恒重的扁形瓶(两次烘干后重量差距小于5mg)(x0),称重(x1),置烘箱105℃条件下烘干至两次称重重量差距小于5mg,计x2。
含固量(%)=(x2-x0)/(x1-x0)×100%
按2.4固含量测定方法,测定不同批次水提取液的固含量,结果如表5所示。
表5水提取过程固含量测定结果
2.5定量模型的建立
选择最优建模的波段和光谱预处理方建立水提取过程芍药苷、固含量定量检测模型。芍药苷校正集样品的近红外预测值与高效液相测定值的相关关系图如图4所示,固含量校正集样品的近红外预测值与测定值的相关关系图如图5所示。从图中可看出近红外预测值与测定值之间相关性良好,测量结果基本趋于一致,所得模型的参数如表6所示。由表6可知,芍药苷与固含量模型校正集相关系数r2分别达0.97686、0.99633,rmsec分别为0.0645、0.0541,此时对应的rmsecv最小。
表6水提取过程定量检测模型参数
2.6水提取过程预测
用所建立的校正模型对第4批提取的样品进行预测,芍药苷验证集样品的近红外预测值与hplc测定值的相关关系图如图6所示,固含量验证集样品的近红外预测值与测定值的相关关系图如图7所示。芍药苷、固含量预测集相关系数r2分别为0.96859、0.99743,可见预测值与对照值之间有很好的相关性,模型有较好的性能。芍药苷、固含量rmsep分别为0.0623、0.0533,芍药苷、固含量的rsep分别为6.83%、3.78%,控制在10%以内,能够满足中药生产过程实时监控分析的精度要求。
水提取过程一煎液相色谱测定与模型预测芍药苷的浓度变化对照图如图8所示,水提取过程一煎固含量测定与模型预测固含量变化对照图如图9所示,水提取过程二煎液相色谱测定与模型预测芍药苷的浓度变化对照图如图10所示,水提取过程二煎固含量测定与模型预测固含量变化对照图如图11所示。
由图8~图11可知,模型所获得的近红外预测值与芍药苷、固含量测定值基本保持一致,其预测值的过程趋势也与实际过程大体相同;样品的成分浓度高低对近红外光谱技术预测性能影响较大,浓度高的预测效果明显好于较低浓度。
芍药苷、固含量校正模型确立后,采用所建方法,nirs完成一次测量只需20秒,而高效液相完成1次芍药苷含量测定至少需要30min,固含量测定则至少若干小时。因此采用近红外光谱分析技术不仅可以实现中药提取过程有效成分含量及固含量的实时监测,而且该检测方法比常规方法更简便、更快速。
实施例2近红外光谱技术用于气滞胃痛颗粒制备过程中提取挥发油过程的研究
1.仪器与试药
1.1主要药材与试剂
枳壳、香附(炙),辽宁华润本溪三药有限公司提供;新橙皮苷对照品(成都曼斯特生物科技有限公司,批号:must-11030701);柚皮苷对照品(成都曼斯特生物科技有限公司,批号:must-11030612);乙腈(merck,germany);磷酸(aladdin);哇哈哈纯净水。
1.2主要仪器与设备
同实施例1项下的1.2内容。
2.方法与结果
2.1提取液样品的采集
将包含枳壳、香附的12kg药材投入容积为40l的多功能提取罐中,按照生产工艺进行提取。为了保证所采集的样品浓度均匀分布,浸泡阶段每10min取样一次,加热阶段中每8min取样一次,沸腾过程中前60min每6min取样一次其余时间每10min取样一次。共进行6个批次提取挥发油实验。所有批次实验完成后共得到198个样本。将第1、2、3、5、6次实验的提取液样品作为校正集,第4次提取样品作为验证集。
2.2近红外光谱的采集
采集气滞胃痛颗粒提取挥发油过程提取液样品的透射光谱:设定光谱的扫描范围4000~10000cm-1,扫描次数为32次,分辨率为8cm-1,以空气为背景,采用5mm光程的圆柱形玻璃比色管,在一个恒定的环境中(温度为25℃,湿度为35%-45%)采集气滞胃痛颗粒提取挥发油过程的提取液透射光谱,每个样品重复测定3次,计算平均光谱。气滞胃痛颗粒提取过程中提取油部分的原始近红外光谱如图12所示。
通过比较tq优化选择波段以及根据相关系数法选择的波段所建立模型的优劣,最终选择5500~6100cm-1和7170~7270cm-1、5500~6100cm-1以及5600~6000cm-1分别作为柚皮苷、新橙皮苷以及固含量的定量检测模型建模波段;并采用一阶、二阶导数处理、平滑、矢量归一、散射效应校正、多元线性校正、小波变换等多种化学计量学方法,本次模型建立采用一阶、二阶导数处理结合s-g或者norris平滑滤波的方法对光谱进行预处理。表7、表8和表9分别为采用原始光谱和不同预处理方法后建立柚皮苷、新橙皮苷及固含量的近红外定量校正模型得到的r2、rmsec、rmsecv值,通过比较不同预处理方法所建立的模型的rmsec和rmsecv值确定预处理方法。
表7不同预处理方法的提取挥发油过程的柚皮苷定量检测模型的参数
表8不同预处理方法的提取挥发油过程的新橙皮苷定量检测模型的参数
表9不同预处理方法的提取挥发油过程的固含量定量检测模型的参数
由表7、表8和表9可知,在比较了不同预处理方法所建立pls校正模型中,柚皮苷以及新橙皮苷的rmsecv值和rmsec值以经一阶导数处理+norris平滑滤波的方法处理后的光谱建模时为最小,固含量的rmsecv值和rmsec值以经一阶导数处理+s-g平滑滤波的方法处理后的光谱建模时为最小。因此,确定采用一阶导数处理+norris平滑滤波的方法对柚皮苷以及新橙皮苷模型光谱进行预处理;采用一阶导数处理+s-g平滑滤波的方法对固含量模型光谱进行预处理;定量模型采用偏最小二乘法(plsr)进行计算。
2.3橙皮苷、柚皮苷的含量测定
将上述采集到的提取液样品离心10min(13000r/min),移取上清液,进行hplc含量测定,hplc色谱条件如下:
色谱柱:agilentzorbaxsb-c18(4.6×250mm,5μm);
流动相:0.1%磷酸水(a)-乙腈(b);进样量:5μl;
流速:1.0ml/min;柱温:30℃;检测波长:283nm;
洗脱梯度程序如表10所示,含量测定结果如表11所示。
表10梯度洗脱程序
表11提取挥发油过程的柚皮苷、新橙皮苷含量测定结果
2.4固含量测定
提取液静置24h,量取4ml上清液至已烘干至恒重的扁形瓶(两次烘干后重量差距小于5mg)(x0),称重(x1),置烘箱105℃条件下烘干至两次称重重量差距小于5mg,计x2。
含固量(%)=(x2-x0)/(x1-x0)×100%
按2.4固含量测定方法,测定不同批次提取挥发油过程的样品液的固含量,固含量测定结果如表12所示。
表12提取挥发油过程的固含量测定结果
2.5定量模型的建立
选择最优建模的波段和光谱预处理方法建立提取挥发油过程中的柚皮苷、新橙皮苷、固含量定量检测模型,所得模型的参数如表13所示。柚皮苷校正集样品的近红外预测值与高效液相测定值的相关关系图如图13所示,新橙皮苷柚皮苷校正集样品的近红外预测值与高效液相测定值的相关关系图如图14所示,固含量校正集样品的近红外预测值与测定值的相关关系图如图15所示。
由图13~图15可知,柚皮苷以及新橙皮苷的近红外预测值与测定值之间相关性良好,测量结果基本趋于一致;固含量的近红外预测值与测定值之间相关性良好,测量结果基本趋于一致。由表13可知,柚皮苷、新橙皮苷及固含量模型校正集相关系数r2分别达0.99302、0.99267、0.99716,rmsec分别为0.269、0.124、0.159,此处对应rmsecv最小。
表13提取挥发油过程的定量检测模型参数
2.6提取挥发油过程预测
用所建立的校正模型对第4批提取的样品进行预测,柚皮苷验证集样品的近红外预测值与高效液相测定值的相关关系图如图16所示,新橙皮苷验证集样品的近红外预测值与hplc测定值的相关关系图如图17所示,固含量验证集样品的近红外预测值与测定值的相关关系图如图18所示。由图16~图18可知,柚皮苷、新橙皮苷、固含量预测集相关系数r2分别为0.99345、0.99843、0.99816,可见预测值与对照值之间有很好的相关性,模型有较好的性能;柚皮苷、新橙皮苷、固含量rmsep分别为0.265、0.162、0.305,柚皮苷、新橙皮苷、固含量的rsep分别为5.71%、7.73%、6.66%,控制在10%以内,能够满足中药生产过程实时监控分析的精度要求。
提取挥发油过程的液相色谱测定与模型预测柚皮苷的浓度变化对照图如图19所示,提取挥发油过程的液相色谱测定与模型预测新橙皮苷的浓度变化对照图如图20所示,提取挥发油过程的固含量测定与模型预测固含量变化对照图如图21所示。由图19~图21可知,模型所获得的近红外预测值与柚皮苷、新橙皮苷、固含量测定值基本保持一致,其预测值的过程趋势也与实际过程大体相同;此外,样品的成分浓度高低对近红外光谱技术预测性能影响较大,浓度高的预测效果明显好于较低浓度。
柚皮苷、新橙皮苷、固含量的校正模型确立后,采用所建方法,nirs完成一次测量只需20秒,而高效液相完成1次柚皮苷、新橙皮苷含量测定至少需要30min,固含量测定则至少若干小时。因此,采用近红外光谱分析技术不仅可以实现中药提取过程有效成分含量及固含量的实时监测,而且其检测方法比常规方法更简便、更快速。
实施例3近红外光谱技术用于气滞胃痛颗粒制备过程中提取液浓缩过程的研究
1、实验材料
1.1药材与试剂
气滞胃痛颗粒浓缩液(辽宁华润本溪三药有限公司提供);芍药苷对照品(成都曼斯特生物科技有限公司,批号:must-11030608);乙腈(merck,germany);磷酸(aladdin);哇哈哈纯净水。
1.2主要仪器及设备
同实施例1项下的1.2内容。
2.方法与结果
2.1浓缩液样品的采集
由于辽宁华润本溪三药生产车间浓缩设备(盘管式真空浓缩锅)在浓缩过程中可以取出浓缩液样品,浓缩过程样品在辽宁华润本溪三药有限公司生产车间现场采集。提取液的静置过滤液在输入盘管式真空浓缩锅并到达预设的浓缩温度和压力时作为计时零点并开始取样,样品体积为50ml。前8小时左右每隔一小时取样。当所有单效和双效中的浓缩液输入到盘管式真空浓缩锅后每隔20分钟取样一次,直至出膏,重复10个批次。所有批次实验完成后共得到120个样本。将第1、2、3、4、6、7、8、9、10次实验的浓缩液样品作为校正集,第5次浓缩液样品作为验证集。
2.2近红外光谱采集
采集气滞胃痛颗粒浓缩液的透射光谱:设定光谱的扫描范围4000~10000cm-1,扫描次数为32次,分辨率为8cm-1,以空气为背景,采用5mm光程的圆柱形玻璃比色管,在一个恒定的环境中(温度为25℃,湿度为35%~45%)采集气滞胃痛颗粒浓缩液透射光谱,每个样品重复测定3次,计算平均光谱。气滞胃痛颗粒浓缩液的原始近红外光谱如图22所示。
采用tq、opus以及相关系数法分别选择了一系列波段,通过比较各个波段以及不同波段组合的建模效果,最后选择5449.8~6101.7cm-1、5149.01~5442.13cm-1和5469.13~7143.04cm-1、5149.01~5442.13cm-1和5469.13~7143.04cm-1分别作为芍药苷、相对密度、固含量的建模波段,并采用一阶、二阶导数处理、平滑、矢量归一、散射效应校正、多元线性校正、小波变换等多种化学计量学方法,本次模型建立采用一阶、二阶导数处理结合s-g或者norris平滑滤波的方法对光谱进行预处理,采用原始光谱和不同预处理方法后建立芍药苷、相对密度、固含量的近红外定量校正模型得到的r2、rmsec、rmsecv值分别如表14、表15和表16所示,通过比较不同预处理方法所建立的模型的rmsec和rmsecv值确定预处理方法。
表14不同预处理方法的提取液浓缩过程芍药苷定量检测模型的参数
表15不同预处理方法的提取液浓缩过程的相对密度定量检测模型参数
表16不同预处理方法的提取液浓缩过程的固含量定量检测模型参数
由表14、表15和表16可知,不同预处理方法所建立pls校正模型中,芍药苷的rmsecv值和rmsec值以经二阶导数处理+norris平滑滤波的方法处理后的光谱建模时为最小,相对密度和固含量的rmsecv值和rmsec值以经一阶导数处理+norris平滑滤波的方法处理后的光谱建模时为最小。因此,采用二阶导数处理+norris平滑滤波的方法对芍药苷模型光谱进行预处理,采用一阶导数处理+norris平滑滤波的方法对相对密度及固含量模型光谱进行预处理;定量模型采用偏最小二乘法(plsr)进行计算。
2.3芍药苷含量测定
将上述采集到的提取液样品离心10min(13000r/min),移取上清液,根据样品浓度用蒸馏水稀释5倍或10倍,进行hplc含量测定,hplc条件如下:
色谱柱:agilentzorbaxsb-c18(4.6×250mm,5μm);
流动相:0.1%磷酸水(a)-乙腈(b);进样量:5μl
流速:1.0ml/min;柱温:30℃;检测波长:230nm;
洗脱梯度程序见表17所示。
表17梯度洗脱程序
按2.3色谱条件分析,进样量为5μl,测定不同批次芍药苷的含量。芍药苷的含量测定结果如表18所示,芍药苷对照品的hplc图谱如图23所示,芍药苷样品的hplc图谱如图24所示。
表18提取液浓缩过程的芍药苷含量测定结果
2.4相对密度测定
浓缩液样品离心(13000r/min,10min)后移取上清液,置于50℃水浴锅中恒温保持30min。精密移取1ml上清液样品,称重,所得数值即为浓缩样品相对密度值(g/ml),按2.4相对密度测定方法,测定不同批次浓缩液的相对密度,结果见表19。
表19提取液浓缩过程的相对密度测定结果
2.5固含量测定
提取液静置24h,量取4ml上清液至已烘干至恒重的扁形瓶(两次烘干后重量差距小于5mg)(x0),称重(x1),置烘箱105℃条件下烘干至两次称重重量差距小于5mg,计x2。
含固量(%)=(x2-x0)/(x1-x0)×100%
按“2.5固含量测定”方法,测定不同批次浓缩液的固含量,含量测定结果如表20所示。
表20提取液浓缩过程的固含量测定结果
2.6定量模型的建立
选择最优建模的波段和光谱预处理方建立提取液浓缩过程的芍药苷、相对密度、固含量定量检测模型,所得模型的参数如表21所示。芍药苷校正集样品的近红外预测值与高效液相测定值的相关关系图如图25所示,相对密度校正集样品的近红外预测值与测定值的相关关系图如图26所示,固含量校正集样品的近红外预测值与测定值的相关关系图如图27所示。由图25~图27可知,近红外预测值与测定值之间相关性良好,测量结果基本趋于一致。由表21可知,芍药苷、相对密度、固含量模型校正集相关系数r2分别达0.98125、0.99551、0.99977,rmsec分别为0.597、0.00584、0.247,此时对应rmsecv最小。
表21提取液浓缩过程的定量检测模型参数
2.7提取液浓缩过程预测
用所建立的校正模型对第5批提取的样品进行预测,芍药苷验证集样品的近红外预测值与hplc测定值的相关关系图如图28所示,相对密度验证集样品的近红外预测值与测定值的相关关系图如图29所示,固含量验证集样品的近红外预测值与测定值的相关关系图如图30所示。由图28~图30可知,芍药苷、相对密度、固含量预测集相关系数r2分别为0.99859、0.99876、0.99743,这表明,预测值与对照值之间有很好的相关性,模型有较好的性能;芍药苷、相对密度、固含量的rmsep分别为0.407、0.00677、0.436,芍药苷、相对密度、固含量的rsep分别为7.10%、0.62%、1.86%,控制在10%以内,能够满足中药生产过程实时监控分析的精度要求。
提取液浓缩过程固含量测定与模型预测固含量的浓度变化对照图如图31所示,提取液浓缩过程相对密度测定与模型预测相对密度的浓度变化对照图如图32所示,提取液浓缩过程固含量测定与模型预测固含量的浓度变化对照图如图33所示。由图31~图33可知,模型所获得的近红外预测值与芍药苷、相对密度、固含量测定值基本保持一致,其预测值的过程趋势也与实际过程大体相同。
芍药苷、相对密度、固含量校正模型确立后,采用所建方法,nirs完成一次测量只需20秒,而高效液相完成1次芍药苷含量测定至少需要30min,相对密度、固含量测定则至少若干小时。因此,采用近红外光谱法不仅可以实现中药提取过程有效成分含量及固含量的实时监测,而且其检测方法比常规方法更简便、更快速。
综上,本发明建立了气滞胃痛颗粒的制备过程中水提取过程、提取挥发油过程和提取液浓缩过程的近红外光谱在线分析方法,并将其应用于在线检测,实现了气滞胃痛颗粒的制备过程的自动化检测和各质控指标含量的实时预测;所建立的模型预测精度高,准确度高,满足实际生产中定量分析的要求。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!