奥泽沙星杂质及其用途的制作方法
本发明涉及一种无氟喹诺酮抗菌药的杂质,具体的说为奥泽沙星的杂质及其应用。
背景技术:
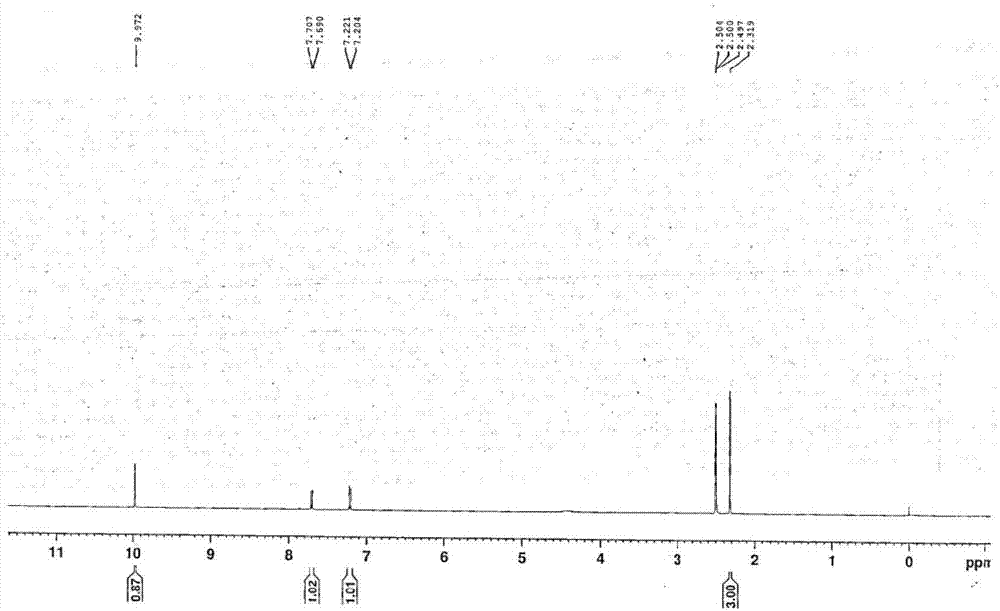
:奥泽沙星(ozenoxacin)是ferrer公司研发的一种用于治疗脓疱性皮炎和其他皮肤感染的新型无氟喹诺酮抗菌药,通过抑制dna旋转酶和拓扑异构酶iv,作为乳膏(10g,1%)治疗皮肤细菌感染,包括革兰氏阳性的皮肤和软组织感染。目前该化合物已经成功完成了治疗成人及儿童脓疱性皮炎的iii期临床试验,显示出更强的抗菌活性,安全性和耐受性均较好。奥泽沙星已于2014年7月在日本上市,2015年9月获批用于治疗痤疮和皮肤感染(10g,2%,凝胶剂)。奥泽沙星的化学合成线路较长,在其合成过程中,式ii化合物是其重要的一个中间体。式ii化合物为已知化合物,在德国专利de3441788(发明名称:alkyl-1-cyclopropyl-1,4-dihydro-4-oxo-3-chinolincarbonsaeuren,verfahrenzuihrerherstellungsowiedieseenthaltendeantibakteriellemittel,1986年5月15日公开)中公开了其制备方法如下:本申请人在合成试验中发现式ii的纯度将直接影响到最终产物奥泽沙星的产品质量。在式ii的药学分析中发现其有一较大杂质峰,这一较大杂质峰经过工艺优化仍不能消除(最低归一含量0.5%),且无法确切推断其来源。对该杂质进行分离制备,通过对其结构确证,以此可以确定其来源,产生机理,在工艺中控制其反应试剂和反应条件(如碳酸钾的添加、水分等),从而防止或降低杂质的产生,进一步控制其含量,保证最终产品奥泽沙星的质量。技术实现要素:本发明的目的提供高纯度的奥泽沙星。本发明提供了一种奥泽沙星中间体杂质,如下式i所示。分子式:c11h7clo4分子量:238.62本发明还提供了一种奥泽沙星中间体杂质的用途,即在检测奥泽沙星原料和/或制剂中的应用。本发明还提供了一种制备奥泽沙星中间体杂质的方法,包括以下步骤:a、称取奥泽沙星中间体,用乙腈-水溶液溶解制成中间体的乙腈-水溶液,b、取步骤a的乙腈-水溶液,注入制备液相色谱仪,c、收集中间体杂质目标馏分,d、连续多次进样,收集合并目标组分,e、旋蒸发浓缩干燥,即得。其中所述的步骤a的中间体浓度为5~30mg/ml,优选为20~30mg/ml。其中所述的步骤a的乙腈-水溶液,乙腈与水的体积比为1:0.10~1,优选为1:0.15~0.35。其中收集馏分的时间大约为1.3-3.5min左右。具体可视a、b流动相的洗脱比例及不同流速做调整。其中所述的色谱条件为:色谱柱以十八烷基硅烷键合硅胶为填充剂;检测波长:240nm;流速:10~30ml/min,进样体积:2~10ml以水、乙腈作为流动相进行梯度洗脱。其中洗脱梯度如下表所示:本发明提供了一种奥泽沙星重要杂质,其纯度达99%以上,为奥泽沙星杂质检测定量及定性分析提供对照品,可用于奥泽沙星原料和/或制剂中杂质对照定位,定性或定量使用,对奥泽沙星用药安全具有非常重要的指导意义。本发明提供的奥泽沙星杂质制备方法,简单方便,易于操作,易于放大批量制备。本发明未涉及到缓冲盐,省去了除盐过程,利于提高收率。本发明可获得高纯度的奥泽沙星杂质。附图说明图1为式i化合物红外测定图图2为式i化合物核磁共振氢谱测定图图3为式i化合物核磁共振碳谱测定图图4为式i化合物hplc检测纯度图具体实施方式实施例1、式ii的制备按德国专利de3441788所述方法,获得式ii化合物。实施例2、式i的制备仪器及试剂吉尔森gx-281制备色谱系统,旋转蒸发仪:申胜旋转蒸发仪laborota4001,色谱柱型号为色谱柱:waterssunfireprepc18obd:以十八烷基硅烷键合硅胶为填充剂,该色谱柱的柱长250mm、内径19mm、粒径5μm。检测波长:240nm;流速:15ml/min,进样体积:9ml流动相a:水;流动相b:乙腈梯度洗脱:时间(min)流动相a(%)流动相b(%)0703057030659511595127030157030称取奥泽沙星中间体样品300mg,即式ii化合物,经高效液相色谱法检测,式i含量约为5%。将8.5ml乙腈与1.5ml水配制成乙腈水溶液,将式ii化合物溶解在的乙腈水溶液中,制成浓度为30mg/ml的溶液。按上述色谱条件进行收集1.7-3.5min时间的馏分,连续多次进样,收集合并目标组分,进行旋蒸发浓缩干燥,得白色固体。式i的结构确证如下:红外:3369cm-1为芳羟基的伸缩振动υ-o-h,1292cm-1为芳羟基的伸缩振动υc–oh,1362cm-1为羟基的变形振动δ-oh;2873cm-1处为甲基的对称伸缩振动υ-c-h,1427cm-1处为甲基的不对称变形振动δ-c-h;2820cm-1处为醛基的伸缩振动υ-c-h,1394cm-1处为醛基的变形振动δ-c-h;1703cm-1、1689cm-1处强的吸收为羰基的伸缩振动υc=o;1633cm-1、1587cm-1、1550cm-1、1474cm-1处吸收峰为苯环骨架振动υc=c;860cm-1、829cm-1处为芳环氢δar-h;1212cm-1处吸收峰为酯反对称伸缩振动υc-o-c;1196cm-1处吸收峰为酯反对称伸缩振动υc-o-c;776cm-1处吸收谱带为单氯代物υc-cl吸收。红外测定结构如图1所示。核磁共振氢谱(1h-nmr)(500mhz,dmso-d6):δ9.97(s,1h),7.70、(d,j=8.5,1h),7.21(d,j=8.5,1h),2.32(s,3h)。核磁共振氢谱测定结构如图2所示。核磁共振碳谱(13c-nmr)(125hz,dmso-d6):δ186.98,δ177.12,δ162.40,δ153.73,δ136.93,δ124.17,δ123.34,δ123.13,δ121.79,δ101.94,δ12.86。核磁共振碳谱测定结构如图3所示。质谱分析结果m/z515.3[2m+k]+,m/z237.0[m-h]-,高分辨质谱分析结果c11h7clo4[m-h]+理论值为:236.9960,实测值为:237.0072,偏差为0.0047%,小于0.005%,在可接受范围内。以上图谱说明所得白色固体即为式i化合物。经hplc检测,式i化合物纯度为99.29%,hplc检测纯度如图4所示。实施例3、式i的制备仪器及试剂与实施例2相同。检测波长:240nm;流速:20ml/min,进样体积:3ml流动相a:乙腈;流动相b:水以与实施例2相同的梯度洗脱。称取奥泽沙星中间体样品2g,即式ii化合物,经高效液相色谱法检测,式i含量约为5%。将75ml乙腈与25ml水配制成乙腈水溶液,将式ii化合物溶解在的乙腈水溶液中,制成浓度为20mg/ml的溶液。按上述色谱条件进行收集1.3-2.7min时间的馏分。连续多次进样,收集合并目标组分,进行旋蒸发浓缩干燥,得白色固体。经红外、核磁共振氢谱、核磁共振碳谱、质谱分析检测,各个图谱与实施例2一致,说明所获得的白色固体为式i化合物。经hplc检测,式i化合物纯度为99.31%。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1