一种测定达巴万星杂质的方法与流程
本发明属于医药分析
技术领域:
,特别涉及一种测定达巴万星中3-二甲氨基丙胺的分析方法。
背景技术:
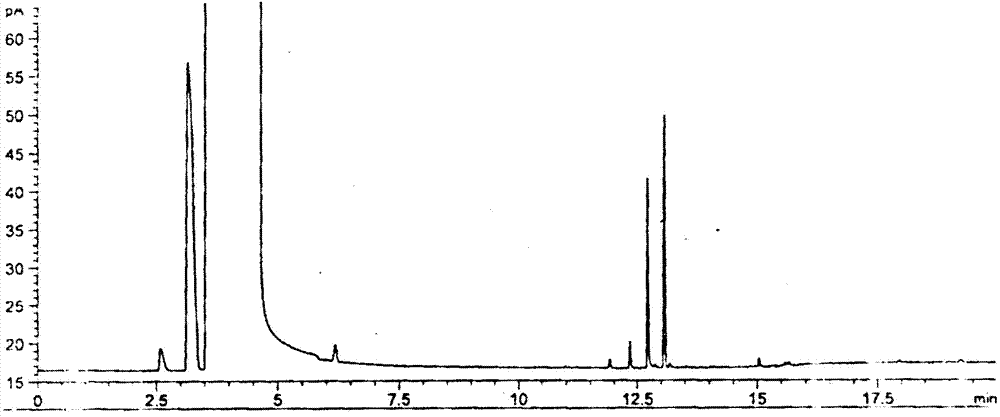
:达巴万星(dalbavancin,结构式如下),也称作道古霉素,它是一种新型半合成糖肽抗生素,为替考拉宁类似物a40926的衍生物。盐酸达巴万星是首个也是唯一一个获批用于absssi治疗的双剂量方案的静脉注射抗生素。达巴万星作用机制与万古霉素和替考拉宁相同,抑制g+菌细胞壁的生物合成,被广泛用作治疗皮肤和软组织感染的药物。达巴万星是第二代、半合成脂糖肽,将一个亲脂性侧链加入到一种增强的糖肽主链,该药在体外对多种革兰氏阳性菌(包括mrsa和化脓性链球菌)及其他链球菌种细菌均表现出杀菌活性。达巴万星具有独特的药动学性质,可每周间隔用药。目前,达巴万星在治疗导管相关的血源性感染以及皮肤和软组织感染已取得了良好的效果,其具有优良的体内抗菌活性和安全性,是理想的第二代糖肽抗生素。达巴万星合成过程中使用3-二甲氨基丙胺作为原料之一,从而在最终成品可能存在3-二甲氨基丙胺的残留。3-二甲氨基丙胺(简称dmapa),n,n-二甲基-1,3-丙二胺,英文名为n,n-dimethyl-1,3-propanediamine,cas登记号为109-55-7,分子式是c5h16n2,分子量为104.1928,无色液体,具有氨味。dmapa密度0.8100g/cm2,凝固点-70℃,沸点134℃,分子量102。dmapa化学性能活泼,能溶于水和有机溶剂,具有较强的挥发性、毒性、腐蚀性,对皮肤和眼睛有较强刺激性,同时含有伯胺基,有产生亚硝胺类或亚硝基化合物的潜在可能。因此,建立合适的达巴万星中3-二甲氨基丙胺含量检测方法对其进行严格控制就十分必要。中国专利《一种测定两性表面活性剂中微量n,n-二甲基丙二胺的分析方法》(申请号20111015766.1)公开了在测定两性表面活性剂中微量n,n-二甲基丙二胺的分析方法:采用离子色谱法进行分析,色谱检测条件如下:色谱柱:阳离子色谱柱;流动相:由体积百分含量为70-100%的无机酸或有机酸酸水溶液和体积百分含量为0-30%的有机溶剂组成,所述无机酸或有机酸水溶液的摩尔浓度为1-10mmol/l;检测器:采用电导检测器;流动相流速:0.5-1.5ml/min。有机溶剂为甲醇或乙腈。此专利所公开的离子色谱法,采用阳离子分离柱、电导检测器分析两性表面活性剂中3-二甲氨基丙胺的含量,方法讨论3-二甲氨基丙胺标准溶液线性情况,没有对3-二甲氨基丙胺在表面活性剂样品中的线性和回收进行研究。采用阳离子分离柱和电导检测器价格昂贵,操作要求高,使用维护成本高。采用液相色谱使用到大量的酸性溶剂和有机溶剂,检测成本高且不够环保。该专利采用酸性溶剂溶解样品测定n,n-二甲基丙二胺,而盐酸达巴万星在酸性溶剂中溶解性差,该专利方法无法适用于盐酸达巴万星中的n,n-二甲基丙二胺的测定。本发明找到一种与其不同原理的气相色谱法,操作简单,通用性强,系统耐用性好,稳定性良好,适合酸性化合物达巴万星中3-二甲氨基丙胺含量检测方法,检测成本低,环保。文献《烷基酰胺丙基甜菜碱中3-二甲氨基丙胺的测定》(夏雄燕等,广东化,2013,40(19)152-153)公开了采用固相萃取柱预处理气相色谱分析方法。这种方法对样品前处理操作复杂,对操作人员要求高,通用性差,不同量添加回收率差异大。此外,此法对测定达巴万星中的3-二甲氨基丙胺不适用,因为达巴万星在所采乙醇-水溶剂体系中溶解性差,难以满足测定要求,而且达巴万星中结合的3-二甲氨基丙胺无法形成游离胺,不能有效萃取和汽化。因药品对最终产物纯度的高要求,3-二甲氨基丙胺的残留含量很低,采用常规分析方法酸碱滴定法测定时,一般滴定法的灵敏度和准确度达不到要求。3-二甲氨基丙胺无紫外吸收波长,有报道采用荧光衍生化液相色谱,荧光检测器进行检测,但此法需对3-二甲氨基丙胺进行荧光衍生处理,操作复杂,前处理繁琐。3-二甲氨基丙胺显碱性,而达巴万星显较强的酸性,故不能采用常规的中性或酸性溶剂溶解后,直接进样气相色谱法测定。3-二甲氨基丙胺与达巴万星结构中众多的酸性基团结合而不能游离汽化,即使采用三乙胺也无法置换游离。盐酸达巴万星及其达巴万星游离碱在众多溶剂中溶解性差,限制了溶剂的选择。达巴万星易溶于氢氧化钠溶液,而氢氧化钠溶液含有大量无机碱,不利于采用气相色谱法溶液直接进样测定。由于3-二甲氨基丙胺具有很强的极性,难于选择合适的溶剂实现有效的液相提取。技术实现要素:本发明为了克服现有技术中达巴万星溶解性差、达巴万星中的3-二甲氨基丙胺无法有效游离汽化、不能有效液相提取等技术缺陷,提供一种简单通用、检测灵敏度高的检测达巴万星中的3-二甲氨基丙胺的方法。本发明所提及的达巴万星,含达巴万星游离碱及达巴万星盐酸盐。本发明提供的检测方法采用强碱溶液对酸性化合物达巴万星进行溶解,并将结合成盐的3-二甲氨基丙胺置换释放成3-二甲氨基丙胺游离碱。利用强极性溶剂乙腈与碱溶液不互溶产生分层的现象,采用乙腈对达巴万星强碱溶液中强极性的3-二甲氨基丙胺进行液-液提取,添加饱和的无机盐增强乙腈的提取效果,采用通用性强的火焰离子化检测器,采用直接进样气相色谱法取乙腈层(上层)进样,从而避免强碱进入色谱系统损坏系统。具体的说,本发明通过下述技术方案得以解决:a、供试品溶液制备:精密称取达巴万星,按质量体积比1:5~50加入浓度为0.5~5mol/l的强碱溶液;溶解后加入乙腈,乙腈与强碱溶液体积比为1:0.5~10;再加入无机盐,无机盐与强碱溶液的质量体积比为1:0.5~10(g/ml);振摇充分混合,静置分层,吸取上层清液,作为供试品溶液,备用;b、对照品溶液制备:精密称取3-二甲氨基丙胺,加入浓度为0.5~5mol/l的强碱溶液,定量稀释制成每1ml中分别含3-二甲氨基丙胺50μg、100μg、150μg的溶液;精密量取上述溶液,加与上述溶液体积比为1:0.5~10的乙腈,加上述溶液质量体积比为1:0.5~10(g/ml)的无机盐,振摇充分混合,静置分层,吸取上层清液,作为对照品溶液(1)~(3),备用;c、以气相色谱法进行检测,按标准曲线法以峰面积计算3-二甲氨基丙胺的含量。其中所述的强碱为氢氧化钠和/或氢氧化钾,优选为氢氧化钠。本发明选择强碱溶液解决了在其他众多溶剂中不溶的问题,保证达巴万星的高溶解性,利于保证高的检测灵敏度。强碱的碱性优于三乙胺,能达到将3-二甲氨基丙胺形成游离碱,从而保证了3-二甲氨基丙胺在气相色谱中能完全汽化。满足回收率在80%~120%之间的要求,故能准确定量检出达巴万星中可能残留的3-二甲氨基丙胺。为了保证样品良好的溶解和3-二甲氨基丙胺良好的游离,强碱的浓度选择为0.5~5mol/l,优选为2~3mol/l。若强碱溶液浓度太低,3-二甲氨基丙胺不能完全游离,平均回收率低,不能准确定量检出达巴万星中可能残留的3-二甲氨基丙胺。其中所述的步骤a中,达巴万星与强碱溶液的质量体积优选为1:10。其中所述的步骤a与b中,乙腈与强碱溶液的体积比优选为1:0.5~2,更优选为1:1。本发明采用了强碱溶液-乙腈不互溶体系提取,形成水相有机相分层,解决了乙腈-水体系互溶的问题。本发明采用乙腈提取,解决了其他弱极性溶剂不能有效提取的问题。同时回避了强碱溶液含有大量无机碱及盐,不利于直接进样的问题。其中所述的无机盐为氯化钠、氯化钾、无水硫酸钠中的任一种或多种。优选为氯化钠。其中所述加入的无机盐与强碱溶液的质量体积比为1:2。其中所述加入的乙腈与无机盐的顺序为先加入乙腈或先加入无机盐。本发明可以采用各种气相色谱仪,比如型号为agilent7890b、色谱柱型号为agilentdb-624:可以是以6%氰丙基苯基-94%二甲基聚硅氧烷为固定液的石英毛细管柱,该石英毛细管柱的柱长30m、内径0.32mm、液膜厚度1.8μm。供试品溶液和对照品溶液分别注入气相色谱仪,该气相色谱柱条件为:柱温:程序升温;进样口温度:230~270℃;火焰离子化检测器温度:280~320℃;载气:氮气;载气流速:0.5~2ml/min;分流比:1:1~30:1,注入气相色谱仪的直接进样体积为1~3μl。作为优选,气相色谱柱条件为:柱温:程序升温方法是初始温度60℃保持8分钟,然后以每分钟20℃的速率升温至220℃,保持8分钟;进样口温度:250℃;火焰离子化检测器温度:300℃;载气:氮气;载气流速:1.5ml/min;分流比:5:1;直接进样体积为2μl。在检测时,取对照品溶液连续进样5次,3-二甲氨基丙胺峰面积的相对标准偏差不得过2.0%,以取对照品溶液(1)~(3)的浓度-峰面积作图,计算线性回归方程,相关系数不得低于0.995。分别精密量取供试品溶液和对照品溶液(1)~(3)注入气相色谱仪,依次记录色谱图。按标准曲线法,以3-二甲氨基丙胺峰面积,计算供试品溶液中3-二甲氨基丙胺的含量。与现有技术相比,本发明选择直接进样解决了顶空进样仪器系统对3-二甲氨基丙胺的吸附残留问题。达巴万星中3-二甲氨基丙胺的测定方法,该方法需要方法学验证,包括空白试验、精密度试验、检出限与定量限、线性试验和回收试验,其中所述测定方法中验证线性试验,包括如下步骤:1)配制2.5mol/l的氢氧化钠溶液,备用。2)精密称取对照品3-二甲氨基丙胺100mg置于100ml容量瓶中,用步骤1)中所述氢氧化钠溶液稀释成1000μg/ml的溶液,混匀,备用。3)分别移取0.1、0.25、0.5、1.0、1.5、2.0ml于10ml量瓶中,用步骤1)中所述氢氧化钠溶液稀释定容,摇匀,分别精密量取1ml该溶液,置具塞试管中,加2.5mol/l的氢氧化钠溶液1ml振摇使溶解,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,吸取上层清液,作为待测溶液,备用。4)分别精密量取步骤3)中样品各2μl进样,记录色谱图。5)以浓度为x轴,峰面积为y轴,作线性回归。其中所述测定方法中验证回收试验步骤如下:1)分别配制3-二甲氨基丙胺的浓度为80μg/ml、100μg/ml、120μg/ml的氢氧化钠溶液;所述氢氧化钠溶液浓度为2.5mol/l;所述3-二甲氨基丙胺的浓度为80μg/ml、100μg/ml、120μg/ml的氢氧化钠溶液为对照品溶液;2)精密称取0.1g达巴万星于具塞试管中,用步骤1)中对照品溶液溶解后,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,量取上层清液2μl进样,记录色谱图;3)另取未加入达巴万星的对照品溶液,同法测定,记录色谱图,按标准曲线法计算回收率。本发明采用采用直接进样法,操作简单,对气相相关设备要求低,规避了顶空进样方式原料药和溶剂消耗量大的缺点,也没有出现直接进样带来的出峰复杂和损坏柱子的缺陷。本发明所采用的常规的毛细管色谱柱和氢火焰气相色谱法,简单通用;本发明提供的测定方法检测灵敏度高,最低检出浓度达5.67μg/ml甚至更低浓度;本发明提供的测定方法具有高回收率,回收率达84.24%。本发明成功的提供了3-二甲氨基丙胺的检测方法,从而有助于提高达巴万星的质量及用药安全。本发明所提供的方法可用于酸性化合物中碱性溶剂的检测,如醋酸盐化合物、盐酸盐化合物以及其他各种酸性化合物中的三乙胺、二乙胺、哌嗪、吡啶或其他碱性溶剂。附图说明图1为实施例1中3-二甲氨基丙胺对照品溶液色谱图。图2为实施例1中达巴万星样品检测色谱图。图3为实施例1中空白溶液色谱图。图4为实施例1中3-二甲氨基丙胺检测限色谱图。图5为实施例1中3-二甲氨基丙胺定量限色谱图。图6为实施例1中3-二甲氨基丙胺的线性关系图。具体实施方式1、仪器:分析天平,型号:mettlertoledoabs135-s;气相色谱仪型号为agilent7890b、色谱柱型号为agilentdb-624:以6%氰丙基苯基-94%二甲基聚硅氧烷为固定液的石英毛细管柱,该石英毛细管柱的柱长30m、内径0.32mm、液膜厚度1.8μm。2、试剂:3-二甲氨基丙胺(试剂纯),氢氧化钠(分析纯),乙腈(色谱纯)。3、样品:达巴万星,批号:20161201、20161202、20161203。下面结合实施例对本发明作进一步阐述,但这些实施例不对本发明构成任何限制。实施例1色谱条件:柱温:程序升温:初始温度60℃保持8分钟,然后以每分钟20℃的速率升温至220℃,保持8分钟。进样口温度:250℃;火焰离子化检测器温度:300℃;载气:氮气;载气流速:1.5ml/min;分流比:5:1。1、检测a、供试品溶液制备:配制2.5mol/l的氢氧化钠溶液,备用;取达巴万星约0.1g,精密称定,置具塞试管中,加配制好的氢氧化钠溶液1ml,振摇使溶解,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,吸取上层清液,作为供试品溶液,备用;b、对照品溶液制备:精密称取3-二甲氨基丙胺100mg,置100ml量瓶中,加配制好的氢氧化钠溶液定量稀释制成每1ml中分别含1000μg的溶液,再用配制好的氢氧化钠溶液定量稀释制成每1ml中分别含50μg、100μg、150μg的溶液,精密量取1ml,置具塞试管中,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,吸取上层清液,作为对照品溶液(1)~(3),备用;c、检测方法:参照中国药典2015年版通则0521测定,采用直接进样气相色谱仪法,取对照品溶液连续进样5次,3-二甲氨基丙胺峰面积的相对标准偏差不得过2.0%,以取对照品溶液(1)~(3)的浓度-峰面积作图,计算线性回归方程,相关系数不得低于0.995。分别精密量取供试品溶液和对照品溶液(1)~(3)各2μl,注入气相色谱仪,依次记录色谱图,3-二甲氨基丙胺对照品溶液色谱图如图1所示,达巴万星样品0161201批检测色谱图如图2所示。按标准曲线法,以3-二甲氨基丙胺峰面积,计算供试品溶液中3-二甲氨基丙胺的含量。2、计算公式:线性回归方程y=ax+b3-二甲氨基丙胺残留量(%)=(yi-b)/a×v/w×100%;其中x:3-二甲氨基丙胺对照品溶液浓度(mg/ml);y:3-二甲氨基丙胺对照品溶液的主峰面积;a:线性回归方程斜率;b:线性回归方程截距;yi:供试品溶液中3-二甲氨基丙胺的峰面积;w:供试品溶液配制时样品的取样量(mg);v:供试品溶液配制体积(ml);3、测定结果按照上述测定方法对三批达巴万星供试品进行检测,结果未检出,见表1。表1:批号3-二甲氨基丙胺含量(%)20161201未检出20161202未检出20161203未检出3、方法学验证3.1空白试验精密量取上述配制好的氢氧化钠溶液1ml,置具塞试管中,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,吸取上层清液,作为空白溶液,空白溶液对残留3-二甲氨基丙胺测定无干扰。空白溶液色谱图如图3所示。3.2精密度试验(1)重复性精密称取3-二甲氨基丙胺适量,加上述配制好的氢氧化钠溶液定量稀释制成每1ml中100μg的溶液,精密量取1ml该溶液,置具塞试管中,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,吸取上层清液,作为待测溶液。按上述色谱条件试验测定,连续进样6针测定,结果6针rsd为1.30%,重复性良好。3-二甲氨基丙胺重复性试验结果见表2。表2:编号1#2#3#4#5#6#平均值rsd峰面积325.94320.95324.56334.83324.33325.54326.031.30(2)重现性精密称取3-二甲氨基丙胺适量,加上述配制好的氢氧化钠溶液定量稀释制成每1ml中约含100μg的溶液,精密量取6份1ml该溶液,置具塞试管中,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,吸取上层清液,作为待测溶液。按上述色谱条件试验测定,结果6份rsd为2.80%,重现性良好。3-二甲氨基丙胺重现性试验结果见表3。表3:编号1#2#3#4#5#6#平均值rsd峰面积300.56309.98316.08313.56305.59328.22312.332.803.3检出限与定量限方法:精密称取3-二甲氨基丙胺100mg,用上述配制好的氢氧化钠溶液为溶剂,稀释成7个不同浓度的溶液,见表4。按上述色谱条件试验,精密量取各浓度下该溶液1ml,置具塞试管中,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,吸取上层清液,作为待测溶液。精密量取各待测溶液溶液2μl,分别注入气相色谱仪,记录色谱图。表4:编号浓度(μg/ml)1#226.82#170.13#113.44#56.705#28.356#11.347#6.8048#5.670实验结果:通过逐级降低浓度法测定,按照s/n=3计算,测得3-二甲氨基丙胺的检出限为5.67μg/ml相当样品中0.0057%的量;检测限色谱图如图4所示。通过逐级降低浓度法测定,按照s/n=10计算,测得3-二甲氨基丙胺的定量限为6.80μg/ml相当样品中0.0068%的量;定量限色谱图如图5所示。3.4线性试验精密称取3-二甲氨基丙胺100mg,置100ml量瓶中,用配制好的氢氧化钠溶液稀释制成1000μg/ml的溶液,备用。分别移取0.1、0.25、0.5、1.0、1.5、2.0ml于10ml量瓶中,用配制好的氢氧化钠溶液稀释定容,摇匀。分别精密量取1ml该溶液,置具塞试管中,加配制好的氢氧化钠溶液1ml振摇使溶解,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,吸取上层清液,作为待测溶液。见表5。分别量取2μl进样,记录色谱图。表5:编号溶液浓度(μg/ml)峰面积1#11.3410.202#28.3550.113#56.70119.614#113.4291.305#170.1457.846#226.8670.34结果:以浓度为x轴,峰面积为y轴,作线性回归。线性方程为y=3.0399x-40.8172,相关系数r=0.9979,3-二甲氨基丙胺在11.34~226.8μg/ml浓度范围内线性关系良好。线性关系曲线图如图6所示。3.5回收试验取3-二甲氨基丙胺的浓度为100μg/ml的氢氧化钠溶液以及其响应80%和120%浓度溶液为对照品溶液。精密量取对照品溶液1ml,并加入0.1g样品,振摇溶解后,加乙腈1ml,加氯化钠0.5g,振摇充分混合,静置分层,吸取上层清液,作为待测溶液,量取2μl注入气相色谱仪,记录色谱图。另取未加入样品的对照品溶液,同法测定,按标准曲线法计算回收率。结果试验平均回收率为84.24%,rsd为2.10%(n=9),回收率在80%~120%之间,rsd小于10%,方法回收率良好。见表6。表6:3-二甲氨基丙胺回收率:实施例2供试品、对照品溶液参照实施例1制备。其区别为本实施例的氢氧化钠溶液浓度为2.0mol/l;乙腈为2ml;无机盐为氯化钾,加入量为2g。色谱条件:柱温:程序升温:初始温度60℃保持8分钟,然后以每分钟20℃的速率升温至220℃,保持8分钟。进样口温度:230℃;火焰离子化检测器温度:280℃;载气:氮气;载气流速:1.0ml/min;分流比:1:1;进样体积为1μl。按照上述条件,检测三批供试品溶液,其色谱图中未检测出3-二甲氨基丙胺峰,经计算,达巴万星中3-二甲氨基丙胺的残留量为0%。实施例3供试品、对照品溶液参照实施例1制备。其区别为本实施例的氢氧化钠溶液10ml,浓度为3.0mol/l;乙腈为1ml;无机盐为无水硫酸钠,加入量为1g。色谱条件:柱温:程序升温:初始温度60℃保持8分钟,然后以每分钟20℃的速率升温至220℃,保持8分钟。进样口温度:270℃;火焰离子化检测器温度:320℃;载气:氮气;载气流速:2.0ml/min;分流比:10:1;进样体积为3μl。按照上述条件,检测三批供试品溶液,其色谱图中未检测出3-二甲氨基丙胺峰,经计算,达巴万星中3-二甲氨基丙胺的残留量为0%。实施例4色谱条件与实施例1相同。采用二甲基亚砜作溶剂直接进样气相色谱法测定3-二甲氨基丙胺回收率试验:取3-二甲氨基丙胺的浓度为100μg/ml的二甲基亚砜溶液以及其响应80%和120%浓度溶液为对照品溶液。精密称取已知3-二甲氨基丙胺残留量的达巴万星0.5g于5ml量瓶中,用3-二甲氨基丙胺对照品溶液溶解并稀释至刻度,摇匀,量取2μl注入气相色谱仪,记录色谱图。另取未加入样品的对照品溶液,同法测定,按标准曲线法计算回收率。结果试验平均回收率为0%,rsd为0%(n=9),结果表明此法3-二甲氨基丙胺与达巴万星成盐无法有效游离并汽化,不能有效测定。见表7。表7:采用二甲基亚砜作溶剂直接进样气相色谱法测定3-二甲氨基丙胺回收率试验:当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1