一种环境水体中六溴环十二烷的快速检测方法与流程
本发明涉及环境水体中有机污染物的快速检测分析技术,特别是涉及一种环境水体中六溴环十二烷的快速检测方法。
背景技术:
六溴环十二烷(hbcds)是一种高溴含量的脂环族添加型阻燃剂,主要用于建筑物和汽车的保温材料、电气和电子设备、合成树脂及橡胶涂层等。随着多溴联苯醚(pbdes)在欧洲和北美地区的禁用,hbcd作为pbdes的替代品之一,使用越来越广泛,2006年全球hbcds的产量为20000吨。在欧洲,hbcd用量仅次于四溴双酚a(tbbpa),在我国,hbcds的生产能力约为7500吨/年,且生产量呈逐年增长趋势。随着hbcds的大量使用,在各环境介质中均有检出,其中英国湖泊、比利时河流及我国太湖等水体中hbcds的浓度达到了ppb级。
六溴环十二烷的辛醇水分配较高,采用传统的液-液萃取、索氏抽提、色谱和固相萃取法等方法,普遍存在溶剂消耗量大、处理时间长、操作步骤多、待测物易损失等特征,并对操作人员和环境造成危害。这极大的制约了相关环境监测工作的开展,限制了对水体中六溴环十二烷残留状况,时空分布规律,迁移转化规律的研究。因此,亟需建立快速、高效、环保的六溴环十二烷的检测方法。
技术实现要素:
本发明针对水体中六溴环十二烷在分析检测方面存在的问题,提供一种快速、简便、所需样品量和有机溶剂用量小的分离富集技术,实现对水体中六溴环十二烷的快速检测技术。本发明与现有技术的区别在于,采用spme-hplc-ms/ms串联技术富集、分离、进样、分析检测为一体,实现了对水体中六溴环十二烷的快速检测。
为了实现上述目的,本发明提供了一种环境水体中六溴环十二烷的快速检测方法,其特征在于:该方法包括以下具体步骤:
步骤1、对待测水样进行过滤去除水体悬浮物,准确移取10ml水样于15ml棕色顶空样品瓶中,加入磁力搅拌子,置于操作平台,采用磁力加热搅拌器搅拌加热;
步骤2、对固相微萃取纤维头进行老化处理,老化过程中,流动相为甲醇;流速0.5ml/min,老化处理时间30min,从而得到附着有60μmpdms/dvb涂层的固相微萃取纤维头。
步骤3、将步骤2老化后的固相微萃取纤维头插入顶空样品瓶中,并浸入样品,推出固相微萃取纤维头,在搅拌速度600~1200r/min,温度20~40℃条件下萃取5~40min,收回固相微萃取纤维头;
步骤4、将固相微萃取(spme)进样针插入与高效液相色谱-质谱/质谱(hplc-ms/ms)联用的固相微萃取-高效液相色谱联用(spme-hplc)接口,推出固相微萃取纤维头,扣紧固定扣;
步骤5、待高效液相色谱(hplc)进样针吸入甲醇样,固相微萃取-高效液相色谱联用仪(spme-hplc)接口的六通阀从负载(load)位置扳到喷射(inject)位置时,开始动态解吸过程,流动相通过解吸室,开始冲洗固相微萃取纤维头,使富集的化合物洗脱下来,并随流动相进入色谱柱和质谱进行分离和检测;
步骤6、解吸结束,将六通阀复位至负载(load)位置,打开固定扣,回拉固相微萃取(spme)手柄推杆使固相微萃取纤维头缩回至穿刺隔垫针中,取下固相微萃取(spme)手柄,润洗固相微萃取纤维头,进行下一次的萃取操作;
步骤7、绘制标准曲线,测定环境水样中六溴环十二烷的含量。
进一步地、步骤1中,采用磁力加热搅拌,温度20~40℃,搅拌速度600~1200r/min。
进一步地、步骤2中,收回萃取纤维固相微萃取纤维头做完一个样品需依次用甲醇,超纯水各洗15s。
进一步地、步骤3中,吸附处理条件为搅拌速度800r/min,温度27℃条件下萃取10min,收回固相微萃取纤维头。
进一步地、步骤5中,检测条件为:
色谱柱:waterscortecstmc18,2.7um;4.6×100mm;
流动相:水、甲醇;
流速0.5ml/min;
柱温:40℃;
质谱条件设定为:
电离方式:电喷雾离子源,负离子模式,三重四级杆质谱检测;
喷雾电压:-3500v;
质谱扫描方式:多反应离子监测(mrm);入口电压ep=-10.00v,碰撞室出口电压cxp=-15v。
进一步地,本发明还提供了环境水体中六溴环十二烷的快速检测方法在检测水体中六溴环十二烷方法的应用。
本方法所述环境水体中六溴环十二烷的前处理分析检测方法与传统方法相比较具有快速高效环保的特点。传统方法需要的前处理时间长,操作繁琐,且耗费大量的有机试剂,待测物易损失或污染。
本发明的优点和积极的效果是:操作简便、快速、高效、灵敏、环保、成本低。可用于江河、湖泊等水体中痕量物质的萃取分析,省去繁杂的实验室前处理,大大缩短了环境水样中六溴环十二烷的检测时间。
附图说明
图1α-hbcd的标准曲线;
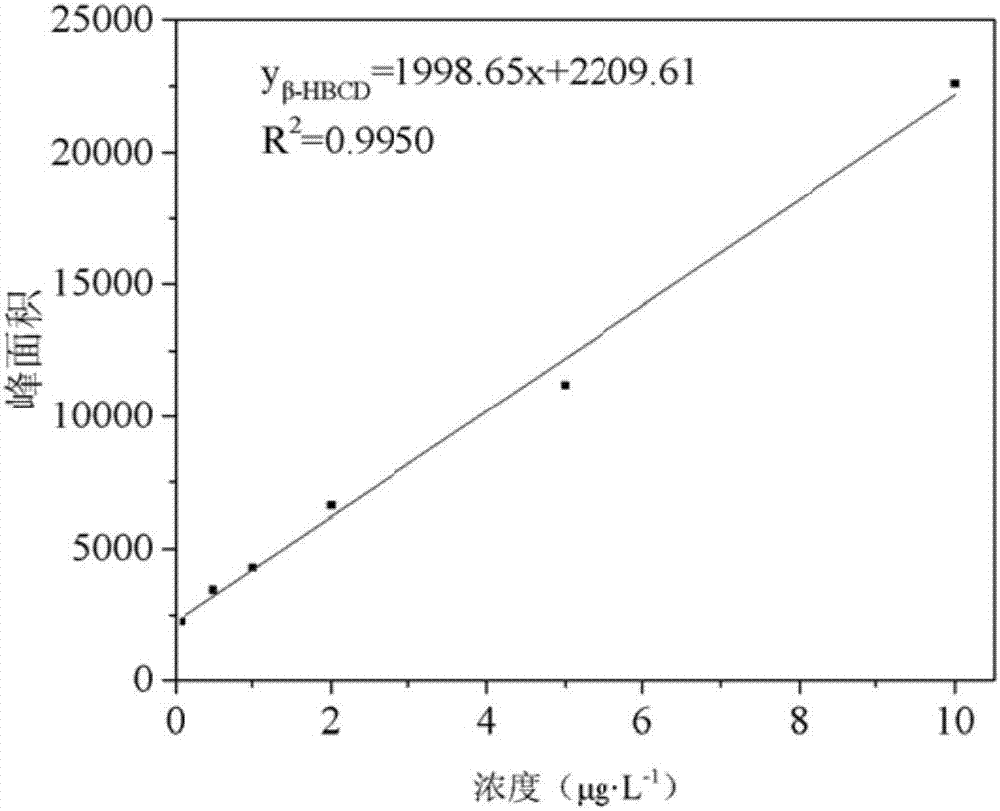
图2β-hbcd的标准曲线;
图3γ-hbcd的标准曲线;
具体实施方式
下面结合实施例和附图1-3对本发明的技术方进行详细说明。
该实施例提供了一种用于水体中六溴环十二烷的快速检测方法,包括以下步骤:
步骤1、对待测水样进行过滤去除水体悬浮物,准确移取10ml水样于15ml棕色顶空样品瓶中,加入磁力搅拌子,置于操作平台,采用磁力加热搅拌器搅拌加热;
该步骤中,采用磁力加热搅拌,温度20~40℃,搅拌速度600~1200r/min。
步骤2、对固相微萃取纤维头进行老化处理,老化过程中,流动相为甲醇;流速0.5ml/min,老化处理时间30min,从而得到附着有60μmpdms/dvb涂层的固相微萃取纤维头。
该步骤中,固相微萃取纤维头做完一个样品需依次用甲醇,超纯水各洗15s。
步骤3、将步骤2老化后的固相微萃取纤维头插入顶空样品瓶中,并浸入样品,推出固相微萃取纤维头,在搅拌速度600~1200r/min,温度20~40℃条件下萃取5~40min,收回固相微萃取纤维头;
优选地,该步骤中,吸附处理条件为搅拌速度800r/min,温度27℃条件下萃取10min,收回固相微萃取纤维头;
步骤4、将固相微萃取(spme)进样针插入与高效液相色谱-质谱/质谱(hplc-ms/ms)联用的固相微萃取-高效液相色谱联用仪(spme-hplc)接口,推出固相微萃取纤维头,扣紧固定扣;
步骤5、待高效液相色谱(hplc)进样针吸入甲醇样,固相微萃取-高效液相色谱联用(spme-hplc)接口的六通阀从负载(load)位置扳到喷射(inject)位置时,开始动态解吸过程,流动相通过解吸室,开始冲洗固相微萃取纤维头,使富集的化合物洗脱下来,并随流动相进入色谱柱和质谱进行分离和检测。
该步骤中,将吸附完成后的固相微萃取纤维头直接进样进行动态解析,利用固相微萃取高效液相色谱质谱联用技术测定水样中的六溴环十二烷,检测条件为:该步骤中,将吸附完成后的固相微萃取纤维头直接进样进行动态解析,利用固相微萃取高效液相色谱质谱联用技术测定水样中的六溴环十二烷,检测条件为:色谱柱:waterscortecstmc18,2.7um;4.6×100mm,即色谱柱规格:长100mm,内径4.6mm,硅胶粒度2.7um;流动相:水、甲醇,见表1;流速0.5ml/min;柱温:40℃;质谱条件设定:电离方式:电喷雾离子源,负离子模式(esi-);三重四级杆质谱检测;喷雾电压:-3500v;质谱扫描方式:多反应离子监测(mrm);入口电压ep=-10.00v,碰撞室出口电压cxp=-15v。碰撞能量见表2。
表1液相色谱梯度洗脱
表2tbbpa定性定量离子
步骤6、解吸结束,将六通阀复位至负载(load)位置,打开固定扣,回拉固相微萃取(spme)手柄推杆使固相微萃取纤维头缩回至穿刺隔垫针中,取下固相微萃取(spme)手柄,润洗固相微萃取纤维头,进行下一次的萃取操作;
步骤7、绘制标准曲线,测定环境水样中六溴环十二烷的含量。
外标法定量时可按常规方式制作标准曲线,对实际样品进行测定,以保留时间定性,以测得目标峰面积值,代入标准曲线方程,求得样品中六溴环十二烷的含量。
表3该实施例所述方法的加标回收率
该实施例中,在本研究优化的测定条件下,对该方法萃取头热稳定性、六溴环十二烷的富集倍数、线性相关系数、检出限、加标回收率及相对标准偏差进行了考察,试验结果表明,萃取头在50℃依然保持良好的热稳定性,六溴环十二烷的富集倍数超过了20倍,使用本发明所述方法对水体中六溴环十二烷的线性范围为0.1~10ng/ml,且线性良好,线性相关系数r2为0.9901~0.9990,加标回收率为81.6%~119.6%,相对标准偏差为2.49%~11.02%,检出限为0.0031~0.013ng/ml,以上数据说明,该方法具有较好的精密度、稳定性和重现性,可以用于水体中六溴环十二烷成分的准确测定。该实施例所述方法的加标回收率如表3所示。
虽然上面结合本发明的优选实施例对本发明的原理进行了详细的描述,本领域技术人员应该理解,上述实施例仅仅是对本发明的示意性实现方式的解释,并非对本发明包含范围的限定。实施例中的细节并不构成对本发明范围的限制,在不背离本发明的精神和范围的情况下,任何基于本发明技术方案的等效变换、简单替换等显而易见的改变,均落在本发明保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!