一种同时测定止咳宁嗽胶囊中三种活性成分的方法与流程
本发明属于医药技术领域,涉及一种中成药止咳宁嗽胶囊中活性成分的检测方法,具体的说是一种切换波长法同时测定止咳宁嗽胶囊中三种活性成分的方法。
背景技术:
止咳宁嗽胶囊收录于《卫生部药品标准·中药成方制剂(第九册)》,由苦杏仁、防风、桔梗、荆芥、陈皮等11味中药组成,具有疏风散寒、宣肺解表、镇咳祛痰的功效,临床用于风寒咳嗽、呕吐、咽喉肿痛等症状。
目前,已有报道采用高效液相色谱法测定止咳宁嗽胶囊中橙皮苷、苦杏仁或橙皮苷与绿原酸的含量。例如:将样品用甲醇超声提取,在zorbaxsb-c18(3.0mm×250mm,5μm)色谱柱上分离,以含1%乙酸的水(a)-乙腈(b)体系为流动相进行阶梯洗脱,程序为0min(82﹕18)~5min(82﹕18)~5.1min(75﹕25)~20min(75﹕25);流速为0.35ml·min-1;327nm紫外检测,可同时测定止咳宁嗽胶囊中的绿原酸和橙皮苷的含量(具体见“高效液相色谱法测定止咳宁嗽胶囊中的绿原酸和橙皮苷”,许海棠等人,广西民族大学化学与生态工程学院,南宁530006)。
然而,上述色谱条件由于流动相中乙酸的浓度过高,测定过程中对仪器和色谱柱会有一定的腐蚀;同时,流速需要控制在0.35ml·min-1,这对仪器的选择也有一定要求,色谱条件过于苛刻。最重要的是,止咳宁嗽胶囊中成分较多,在上述色谱条件下,绿原酸与橙皮苷的色谱峰之间仍存在没有被完全分开的单一的样品峰,无法将止咳宁嗽胶囊中其它活性成分有效的分离出来。流动相组成在5分钟左右由(82﹕18)快速变成(75﹕25),容易引起基线飘移。
为有效监控药品质量,确保该制剂的疗效,需要进一步完善高效液相色谱法,保证其能够快速准确的分析出多种活性成分的含量。
技术实现要素:
本发明目的在于提供一种利用高效液相色谱切换波长法同时测定止咳宁嗽胶囊中的绿原酸、金丝桃苷和橙皮苷含量的方法,为判断该药的质量标准提供理论依据。该方法操作简单、稳定、重现性好,对仪器和色谱柱不会产生不良影响,可用于该药品的质量控制。
本发明的目的是这样实现的:该测定方法是利用高效液相色谱(hplc)切换波长法在同一实验条件下同时分离并测定止咳宁嗽胶囊中的绿原酸、金丝桃苷和橙皮苷的含量,具体步骤如下:
(1)混合对照品溶液的制备:精密称取绿原酸0.00234g、橙皮苷0.00230g和金丝桃苷0.00305g,分别置于5ml容量瓶中,用甲醇溶解并定容至刻度线处;制得浓度分别为468μg·ml-1、460μg·ml-1和610μg·ml-1的标准品溶液;分别移取每种标准品溶液150μl混合,制得混合对照品溶液;
(2)供试品溶液的制备:取16粒止咳宁嗽胶囊,除去胶囊外壳,精密称取4.18535g,用80ml甲醇为溶剂进行索式提取5h,将提取液冷却至室温后转移到100ml容量瓶中,甲醇定容,作为供试品溶液,供检测使用;
(3)分离检测:色谱柱:tc-c18(4.6mm×250mm,5μm);流动相:a为乙腈,b为0.2%冰乙酸的水溶液梯度洗脱,梯度洗脱条件为:
流速:1.0ml·min-1;检测器:vwd检测器;检测波长:0-10min:327nm;10-48min:280nm;柱温:25℃;进样量:10μl。
本发明具有以下优点和积极效果:
1、本发明方法采用高效液相色谱法,利用梯度洗脱方式,实现了同时测定止咳宁嗽胶囊中三种有效成分;能够较好的分离、测定止咳宁嗽胶囊中绿原酸、金丝桃苷和橙皮苷有效成分的含量,色谱峰峰形好,分离度较高,在此色谱条件下止咳宁嗽胶囊中绿原酸、金丝桃苷和橙皮苷的浓度与峰面积呈现良好的线性关系。
2、本发明所述方法操作简单、精密度、准确度、重现性和稳定性好,能够更好地对该药物进行质量监控,为全面评价和控制止咳宁嗽胶囊质量和药效评价的进一步研究奠定了理论基础和科学依据。
附图说明
图1为本发明混合对照品溶液的色谱图。
图2为本发明实施例1供试品溶液的色谱图。
图3为本发明线性关系曲线图。
图4为本发明对比例1混合对照品溶液的色谱图。
图5为本发明不同提取方法下供试品溶液的色谱图。
图6为本发明不同流动相条件下绿原酸的色谱图。
具体实施方式
以下结合附图和具体实施方式对本发明作以进一步的阐述,以便本领域的技术人员更了解本发明,但并不以此限制本发明。
仪器与试药
仪器:agilent1100高效液相色谱仪(安捷伦科技有限公司);uv-2550型紫外分光光度计(日本岛津有限公司);bt-125d十万分之一分析天平(赛多利斯科学仪器有限公司);gwa-un4超纯水器(北京普析通用仪器有限责任公司);sk-5200h超声波清洗器(上海科导仪器有限公司);恒温水浴锅(天津泰斯特仪器公司);shz-d型循环水真空泵(上海华光仪器仪表厂);微孔滤膜(0.45μm)。
试药:乙腈(gr);冰乙酸(ar);甲醇(gr);止咳宁嗽胶囊(吉林吉春制药股份有限公司,批号:z22021693);绿原酸对照品(中国食品药品检定研究院,批号:110753-200413);金丝桃苷对照品(中国食品药品检定研究院,批号:111521-201406);橙皮苷对照品(中国食品药品检定研究院,批号:110721-201316);超纯水。
实施例1
(1)混合对照品溶液的制备:精密称取绿原酸0.00234g、橙皮苷0.00230g和金丝桃苷0.00305g,分别置于5ml容量瓶中,用甲醇溶解并定容至刻度线处;制得浓度分别468μg·ml-1、460μg·ml-1和610μg·ml-1的标准品溶液;分别移取每种标准品溶液150μl混合,制得混合对照品溶液;
(2)供试品溶液的制备:取16粒止咳宁嗽胶囊,除去胶囊外壳,精密称取4.18535g,用80ml甲醇为溶剂进行索式提取5h,将提取液冷却至室温后转移到100ml容量瓶中,甲醇定容,作为供试品溶液,供检测使用;
(3)色谱条件:色谱柱:tc-c18(4.6mm×250mm,5μm);流动相:a为乙腈,b为质量比为0.2%的冰乙酸水溶液梯度洗脱(见表1);流速:1.0ml·min-1;检测器:vwd检测器;检测波长:0-10min:327nm;10-48min:280nm;柱温:25℃;进样量:10μl。
表1梯度洗脱条件
在上述色谱条件下,混和对照品和供试品溶液的色谱图为图1和图2。绿原酸、金丝桃苷和橙皮苷在混合标准品中的保留时间分别是8.367min、16.150min和14.271min。
对比例1待测组分的选择
(1)混合对照品溶液的制备:精密称取绿原酸、升麻素苷、金丝桃苷、5-o-甲基维斯阿米醇、橙皮苷、柚皮苷和连翘苷,分别置于5ml容量瓶中,用甲醇溶解并定容至刻度线处,制得标准品溶液,分别移取每种标准品溶液150μl混合,制得混合对照品溶液。
(2)供试品溶液的制备:参照实施例1。
(3)色谱条件:色谱柱:tc-c18(4.6mm×250mm,5μm);流动相:a为乙腈,b为体积比为0.2%的冰乙酸水溶液水梯度洗脱(见表2);流速:1.0ml·min-1;检测器:vwd检测器;检测波长:263nm;柱温:25℃;进样量:10μl。
表2梯度洗脱程序
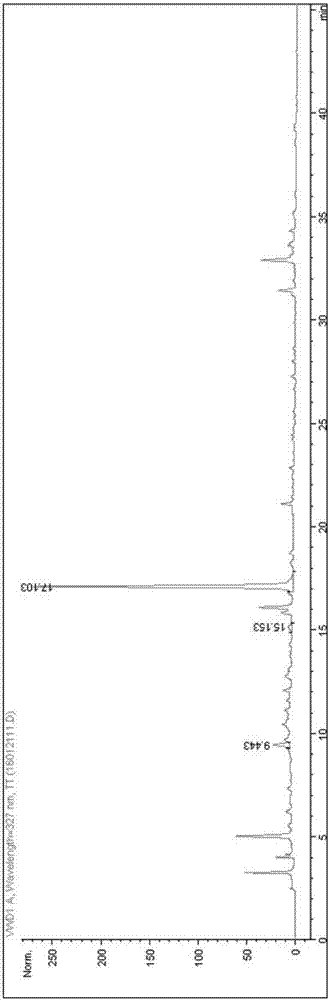
在上述色谱条件下,混和对照品溶液的色谱图为图4,从左到右依次为绿原酸、升麻素苷、金丝桃苷、5-o-甲基维斯阿米醇、橙皮苷、柚皮苷和连翘苷;保留时间分别为3.033min、10.752min、15.556min、19.302min、20.102min、20.366min和22.910min。供试品溶液中升麻素苷、5-o-甲基维斯阿米醇和连翘苷含量较少,用该方法并未检测到上述几种成分。另外,供试品溶液中柚皮苷含量甚微且与橙皮苷较难分离;为保证分析质量,最终确定将绿原酸、金丝桃苷和橙皮苷作为研究对象,进行定性和定量分析。
对比例2供试品溶液制备时提取方法的选择
考察不同提取方法(超声法、浸泡法和索式提取法)对止咳宁嗽胶囊中绿原酸和橙皮苷含量的影响。
(1)超声提取法
取4粒止咳宁嗽胶囊,除去外壳,取胶囊中的粉末精密称取1.04554g,放入锥形瓶中,加入适量的甲醇,超声30min,静置后过滤,将滤液转移到25ml容量瓶中,甲醇定容,作为供试品溶液,供检测使用。
(2)浸泡提取法
取4粒止咳宁嗽胶囊,除去外壳,取胶囊中的粉末精密称取1.04565g,放入锥形瓶中,加入适量的甲醇,浸泡24h,静置后过滤,将滤液转移到25ml容量瓶中,甲醇定容,作为供试品溶液,供检测使用。
(3)索式提取法
取16粒止咳宁嗽胶囊,除去胶囊外壳,精密称取4.18535g,用80ml甲醇为溶剂进行索式提取5h,将提取液冷却至室温后转移到100ml容量瓶中,甲醇定容,作为供试品溶液,供检测使用。
上述超声法提取的供试品溶液的色谱图见图5中的(a)、浸泡法提取的供试品溶液的色谱图见图5中的(b)、索式提取法提取的供试品溶液的色谱图见图5中的(c),不提取方法对止咳宁嗽胶囊中绿原酸和橙皮苷含量的影响见表3。
表3不同提取方法对绿原酸和橙皮苷含量的影响
超声提取法比其它两种提取方法较方便快捷,但是提取的效果不如其它两种提取方法好,可能会影响最后药品含量的检测结果。而索式提取法操作相对繁琐,但是提取的效果最好。浸泡法的操作比较简单,用时较长,提取效果在其它两种提取方法之间。综合考虑,本发明选择索式提取法。
对比例3流动相的选择
由于绿原酸不稳定,在加酸的条件下才能使其稳定存在,为了更好地检测绿原酸,需要在流动相中加酸进行实验。为了确定加何种酸以及加酸量,流动相分别以乙腈~0.05%磷酸、乙腈~0.1%磷酸、乙腈~0.2%磷酸为流动相和乙腈~0.05%冰醋酸、乙腈~0.1%冰醋酸、乙腈~0.2%冰醋酸进行实验,其他色谱条件同实施例1,结果如图6所示,图6中(a)为不加酸;(b)为乙腈~0.05%磷酸;(c)为乙腈~0.1%磷酸;(d)为乙腈~0.2%磷酸;(e)为乙腈-0.05%冰醋酸;(f)为乙腈~0.1%冰醋酸;(g)为乙腈~0.2%冰醋酸。
结果:以乙腈~0.2%冰醋酸(0~15min:10%~30%a;15~30min:30%~85%a;30~40min:85%~85%a;40~48min:85%~10%a)作为流动相进行梯度洗脱的时,色谱峰分离较好,且峰形很好,满足分析的要求。
实施例1色谱条件方法学考查
精密度实验:准确吸取混合对照品溶液,在所述色谱条件下连续进样5次,测定时间为48min。利用绿原酸、橙皮苷和金丝桃苷的峰面积计算相对标准偏差分别为2.67%、3.58%和1.98%(见表4)。结果表明仪器精密度良好。
表4精密度实验数据
重现性实验:取批号相同的止咳宁嗽胶囊5份,供试品溶液的制备方法处理样品,在所述色谱条件下进行测定。绿原酸、橙皮苷和金丝桃苷峰面积的相对标准偏差分别为5.74%、2.98%和3.71%(见表5)。结果表明该法重复性良好。
表5重复性实验数据
稳定性试验:取同一样品溶液,按照所述色谱条件分别测定0、2、4、6、8、10、12和24h的色谱图。根据所得绿原酸、橙皮苷和金丝桃苷的峰面积,计算相对标准偏差分别为3.97%,2.05%和3.05%(见表6)。实验数据表明样品溶液在24h内基本稳定。
表6稳定性实验数据
回收率试验:精密称取样品2份,分别置于容量中,并计算出三种活性成分的含量,以80%的比例在每份样品中均加入对应且适量的绿原酸、橙皮苷和金丝桃苷对照品,混匀后按供试品溶液的测定方法进行含量测定,并计算三种活性成分的平均回收率。再以100%、120%的比例进行加标回收实验,所得数据见表7-表9。
表7绿原酸回收率实验数据
表8橙皮苷回收率实验数据
表9金丝桃苷回收率实验数据
线性关系及含量测定:取对照品溶液,分别在所述色谱条件下进行测定。以浓度为横坐标,色谱峰面积为纵坐标,做峰面积-浓度关系曲线,如图3,图3中(a)为绿原酸,(b)为橙皮苷,(c)为金丝桃苷。三种活性成分回归方程见表10。根据色谱峰面积及线性关系方程计算可得止咳宁嗽胶囊中绿原酸、橙皮苷和金丝桃苷分别为0.16mg·g-1,4.08mg·g-1和0.04mg·g-1。
表10线性关系方程
综上,本发明所建立的高效液相色谱法同时测定止咳宁嗽胶囊中三种活性成分的含量,具有操作简便、精密度好、重复性高的特点,可全面、更有效地控制该药的质量。
- 还没有人留言评论。精彩留言会获得点赞!