一种石榴皮的微提取方法与流程
本发明属于天然药物提取检测领域。主要涉及石榴皮中多酚的提取和测定技术,具体涉及以碳分子筛作为吸附剂的微量基质固相分散技术联合超高效液相色谱串联四级杆飞行时间质谱的测定方法。
背景技术
碳分子筛是一种微孔均匀分布的无定形碳基材料,具有高度发达的孔隙结构,其有效直径在纳米级别。与其他已知的分子筛材料相比,碳分子筛在某些条件(ph,惰性气氛)下具有良好的化学和热稳定性和高疏水性。其高吸附能力和动力学选择性不仅取决于孔的尺寸、形状和微孔体积,也取决于碳分子筛的其它特征,例如微孔入口的尺寸和电子性质。在实践生产中,碳分子筛是由各种碳前体,如煤、碳纤维、核桃壳、树脂、木材等制成。其生产方法主要有两种:一种是基于碳前体的热解,另一种是通过碳沉积技术改性活化结构的孔隙率。近年来,碳分子筛被广泛应用于气体分离、混合物纯化和催化剂或催化剂载体,如从气体中分离o2/n2和co2/ch4等。然而,据我们所知,在微萃取中碳分子筛作为分散剂尚未有相关报道。
基质固相分散(mspd)是一种先进的提取和清理技术,由barker等人提出。mspd可以将样品的提取,分离和净化合并到单个步骤中,已成功应用于各种固体,半固体和液体基质如肝、鱼、牛奶、水果和植物材料中。然而,传统的mspd方法需要消耗大量的时间,样品和溶剂,这对环境并不友好。近年来,微型基质固相分散已经被开发并应用于食品和环境中。与常规的mspd方法相比,mspd微萃取方法具有使用相对少量的样品和溶剂,并且方便的优点。在mspd微萃取方法中,吸附剂的选择是一个关键因素。然而,mspd中经常使用的吸附剂如c18,二氧化硅,氧化铝和弗罗里等在一定程度上缺乏选择性,这将导致杂质和目标分析物的共同提取。因此,应当寻找对mspd微萃取方法具有更广泛适用性和更好选择性的新型吸附剂。
石榴是石榴科石榴属浆果,在世界各地均有栽培。过去几十年来,人们对这种植物药材食品的关注不断提高,因为其既可作为可食用水果,又有助于预防和治疗疾病。到目前为止,已经研究了石榴提取物的各种药理学性质,例如抗菌、消炎、抗氧化、保护心脏和抗糖尿病等。石榴皮是石榴产业的副产品,是一种常用中药,适用于结肠癌、食管癌、肝癌、肺癌和皮肤病等疾病的预防和治疗。石榴皮的这些潜在的功效归因于其具有高含量的多酚类化合物,主要包括可水解的单宁(安石榴林,安石榴苷,烟酸,鞣花酸)和没食子酸等。目前,石榴皮提取物已被广泛应用于食品,化妆品和医药领域。文献中对于石榴皮的检测方法包括液相色谱法、毛细管电泳、高效液相色谱法、高效液相色谱和质谱联用等。在这些测定方法中,样品制备是至关重要的一步。多年来,常规提取技术是使用有机溶剂如甲醇,乙醇等进行超声辅助提取。然而,所述的常规方法不仅耗时,并且需要使用较大体积的样品和有害溶剂。因此,很有必要开发一种更具成本效益和环境友好的样品制备方法。
本发明开发了一种简单,快速和高效的mspd微萃取方法,结合超高效液相色谱四级杆飞行时间质谱(uhplc-q-tof/ms)。以碳分子筛作为mspd微萃取分散剂,改善石榴皮中的多酚(没食子酸,安石榴苷a,安石榴苷b,儿茶素和鞣花酸)的提取效率。评估了几个样品预处理参数以优化萃取过程,包括分散剂的类型,分散剂的量,研磨时间和洗脱溶剂。所开发的方法被验证并应用于从不同地区收集的石榴皮样品的分析和与常规方法的比较。
技术实现要素:
本发明针对传统石榴皮药材提取技术和检测方法中存在的处理过程复杂、耗时、有机溶剂用量大等问题,提出一种以碳分子筛为分散剂的基质固相微萃取与超高效液相色谱串联四级杆飞行时间质谱提取和测定石榴皮中多酚化合物的方法。
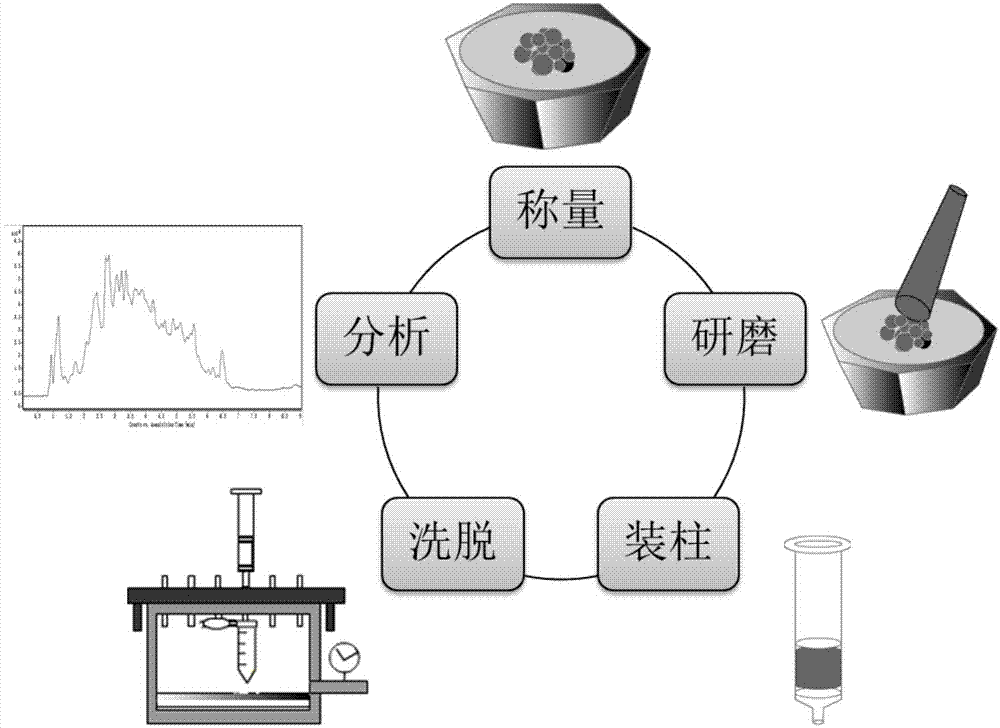
本方法具体是按照以下步骤完成的:
(1)精密称取的26mg干燥的石榴皮样品和一定量的碳分子筛置于玛瑙研钵中,然后用研棒连续搅拌一定时间,直到获得均匀混合物。在完全分散后,将均匀混合物转移到底部装有筛板堵塞的spe柱体中,然后在均匀混合物的顶部加入第二块筛板,压实。用200μl的洗脱溶剂洗脱样品混合物,并通过真空泵保持恒定的流速,将洗脱液收集在1.5ml的离心管中。最后,将洗脱液在离心仪中以13000rpm离心5分钟,取上层清夜,用uhplc-q-tof/ms进行分析。
(2)将各对照品(没食子酸、安石榴苷a、安石榴苷b、儿茶素、鞣花酸)色谱峰的峰面积对其相应浓度进行回归分析,以对照品溶液的浓度为横坐标,谱图中色谱峰的峰面积为纵坐标,制作标准曲线,其线性相关系数大于0.990。将石榴皮样品提取液色谱图中各个目标分析物的峰面积,代入标准曲线,计算得到提取液中各成分的含量。
其中(1)中所述吸附剂类型包括:碳分子筛、c18、介孔碳、硅胶、碳纳米管、氧化石墨烯、弗罗里,优选为碳分子筛。
所述吸附剂的质量为:6.5mg、19.5mg、32.5mg、45.5mg、58.5mg、71.5mg,优选为32.5mg。
所述研磨时间为:30s、60s、90s、120s、150s,优选为90s。
所述洗脱液包括:甲醇、乙醇、丙酮、乙腈、水、乙酸乙酯、甲醇:水(90:10)、甲醇:水(80:20)、甲醇:水(70:30),优选为甲醇。
本发明的优势在于:本发明首次将碳分子筛作为基质固相微萃取的分散剂,有利于对石榴皮中目标分析物的提取。在基质固相微萃取中将样品的萃取和净化合并到单个步骤,提供相对少量的样品和溶剂,快速,经济,符合绿色化学的要求。使用uhplc-q-tof/ms,实现了对石榴果皮中多酚的高灵敏度和选择性分析。
附图说明
图1为本发明基质固相分散萃取方法的流程图。
图2为实施例1考察吸附剂种类对提取效果影响的柱状图。图中,1、2、3、4、5分别代表没食子酸、安石榴苷a、安石榴苷b、儿茶素、鞣花酸。
图3为实施例2考察不同吸附剂质量对提取效果影响的折线图。图中,1、2、3、4、5分别代表没食子酸、安石榴苷a、安石榴苷b、儿茶素、鞣花酸。
图4为实施例3考察不同研磨时间对提取效果影响的折线图。图中,1、2、3、4、5分别代表没食子酸、安石榴苷a、安石榴苷b、儿茶素、鞣花酸。
图5为实施例4考察洗脱溶剂种类对提取效果影响的柱状图。图中,1、2、3、4、5分别代表没食子酸、安石榴苷a、安石榴苷b、儿茶素、鞣花酸。
图6a为五种多酚混合对照品的uhplc-q-tof/ms总离子流图和提取离子流图,b为石榴皮样品提取液的总离子流图和提取离子流图。图中,1、2、3、4、5分别代表没食子酸、安石榴苷a、安石榴苷b、儿茶素、鞣花酸。
具体实施方式
下面结合具体实施例,对本发明作进一步说明。
由于其应用范围广,以下所述实施例是说明性,不是限定性的。
实施例1
精密称取7份26mg干燥的石榴皮粉末分别置于7组研钵中,并称取不同种类的吸附剂(碳分子筛,c18,介孔碳,硅胶,碳纳米管,氧化石墨烯,弗罗里)各32.5mg,分别加入7组研钵中与石榴皮粉末混合研磨90s。取7支1ml规格固相萃取小柱,底部均加入筛板,分别用研磨好的固体混合物填充小柱,顶部均加入筛板,压实后置于固相萃取仪上。用0.2ml的甲醇对小柱进行洗脱,底部用1.5ml规格离心管收集洗脱液。最后,将洗脱液在离心仪中以13000rpm离心5分钟,取上层清夜,用uhplc-q-tof/ms进行分析。
本实验采用使用具有调节自动取样器的安捷伦超高效液相色谱串联四级杆飞行时间质谱(uhplc-q-tof/ms)检测器。
仪器:uhplc1290在agilentsb-c18柱(1.8μm,50mm×4.6mmi.d.)上进行分离分析。用0.1%甲酸水(流动相a)和甲醇(流动相b)梯度洗脱该柱。梯度程序如下:0-2min,5-20%b;2-4min,20-40%b;4-6min,40-60%b;6-8min,60-80%b;8-9min,80-100%b。柱温35℃,洗脱流速设定为0.5ml/min,注射体积为2μl。质谱检测在6530串联四级杆飞行时间质谱仪进行,通过电喷雾电离界面在负离子化模式下连接到uhplc。质谱条件设定如下:气体温度350℃;干燥气流速12l/min;雾化器压力45psig;毛细管电压为3500v;四级杆电压65v;裂解电压为175v,八极杆电压为750v。使用氮气作为雾化器和辅助气体。将碰撞诱导解离(cid)的能量设定为10,20和30ev,并且在100至1500m/z的范围内记录质谱用于定性分析。
七种吸附剂的提取效率柱状图如图2所示,其中碳分子筛实现了目标化合物提取效率的最佳结果。可能碳分子筛具有较大的比表面积和均匀的微孔通道,建立了高吸附能力,对于分散目标化合物是理想的选择。
实施例2
精密称取6份26mg干燥的石榴皮粉末分别置于6组研钵中,并称取不同质量的吸附剂(6.5mg,19.5mg,32.5mg,45.5mg,58.5mg,71.5mg),分别加入6组研钵中与石榴皮粉末混合研磨90s。取6支1ml规格固相萃取小柱,底部均加入筛板,分别用研磨好的固体混合物填充小柱,顶部均加入筛板,压实后置于固相萃取仪上。用0.2ml的甲醇对小柱进行洗脱,底部用1.5ml规格离心管收集洗脱液。最后,将洗脱液在离心仪中以13000rpm离心5分钟,取上层清夜,用uhplc-q-tof/ms进行分析。
不同吸附剂质量对提取效率的影响如图3所示,当碳分子筛的质量从6.5mg增加到32.5mg时,分析物的提取效率随之增加,这归因于分散剂和目标化合物之间的接触面积增加,从而影响吸附能力。当分散剂增加质量为从32.5mg增加到45.5mg时,样品的提取效率略有变化,但并不明显。此时,碳分子筛的吸附能力达到饱和。然而,随着分散剂用量的进一步增加,提取效率反而下降,这可能是因为碳分子筛的过强吸附增强了洗脱过程的难度。
实施例3
精密称取5份26mg干燥的石榴皮粉末分别置于5组研钵中,并称取5份32.5mg的碳分子筛,分别加入5组研钵中与石榴皮粉末混合研磨不同的时间(30s,60s,90s,120s,150s)。取5支1ml规格固相萃取小柱,底部均加入筛板,分别用研磨好的固体混合物填充小柱,顶部均加入筛板,压实后置于固相萃取仪上。用0.2ml的甲醇对小柱进行洗脱,底部用1.5ml规格离心管收集洗脱液。最后,将洗脱液在离心仪中以13000rpm离心5分钟,取上层清夜,用uhplc-q-tof/ms进行分析。
不同研磨时间对提取效果的影响如图4折线图所示,在30s至150s的时间间隔内进行研究。当研磨时间从30s增加到90s时,目标分析物的提取效率显著提高。可能是样品和吸附剂之间的接触面积的增加,促进了样品基质的充分保留。然而,将研磨时间从90s进一步增加到150s时,由于吸附剂的强烈吸附带来了洗脱过程的困难,样品的提取效率反而降低。
实施例4
精密称取7份26mg干燥的石榴皮粉末分别置于7组研钵中,并称取32.5mg碳分子筛加入7组研钵中与石榴皮粉末混合研磨90s。取7支1ml规格固相萃取小柱,底部均加入筛板,分别用研磨好的固体混合物填充小柱,顶部均加入筛板,压实后置于固相萃取仪上。分别用0.2ml的甲醇,90%甲醇,80%甲醇,70%甲醇,乙醇,丙酮,乙腈和乙酸乙酯对小柱进行洗脱,底部用1.5ml规格离心管收集洗脱液。最后,将洗脱液在离心仪中以13000rpm离心5分钟,取上层清夜,用uhplc-q-tof/ms进行分析。
不同洗脱溶剂对提取效果的影响如图5所示。结果显示,甲醇的提取效率最佳。这是由于多酚包含多个酚羟基,根据相似相溶原理,洗脱溶剂极性越强提取效果越好。虽然多酚具有较强的亲水性,但甲醇和水分子作用力的影响,可造成多酚溶解性能的改变。
日内精密度
称取没食子酸,安石榴苷,儿茶素,鞣花酸对照品适量,精密标定,分别置于量瓶中,分别加甲醇制成每lml含1000μg的对照品溶液。将制得的对照品溶液配置成50μg/ml的混合标准溶液,用uhplc-q-tof/ms进行分析。该样品在同一天内连续进样6次。
日间精密度
称取没食子酸,安石榴苷,儿茶素,鞣花酸对照品适量,精密标定,分别置于量瓶中,分别加甲醇制成每lml含1000μg的对照品溶液。将制得的对照品溶液配置成50μg/ml的混合标准溶液,用uhplc-q-tof/ms进行分析。将该样品连续进样3天,每天2次。结果如表1所示。
表1日内、日间精密度实验结果汇总
重复性考察
精密称取26mg干燥的石榴皮粉末分别置于研钵中,并称取32.5mg碳分子筛加入研钵中与石榴皮粉末混合研磨90s。取1ml规格固相萃取小柱,底部均加入筛板,分别用研磨好的固体混合物填充小柱,顶部均加入筛板,压实后置于固相萃取仪上。用0.2ml的甲醇对小柱进行洗脱,底部用1.5ml规格离心管收集洗脱液。最后,将洗脱液在离心仪中以13000rpm离心5分钟,取上层清夜,用uhplc-q-tof/ms进行分析。按上述步骤重复做三组,作为重复性考察。
标准曲线
称取没食子酸,安石榴苷,儿茶素,鞣花酸对照品适量,精密标定,分别置于量瓶中,分别加甲醇制成每lml含1000μg的对照品溶液。将制得的对照品溶液配置成一系列不同浓度的混合标准溶液,用uhplc-q-tof/ms进行检测。将各对照品色谱峰的峰面积对其相应浓度进行回归分析,以对照品溶液的浓度为横坐标,谱图中色谱峰的峰面积为纵坐标,分别制作标准曲线。
没食子酸、安石榴苷a、安石榴苷b、儿茶素、鞣花酸浓度均为50μg/ml的混合对照品总离子流图和提取离子流图如图6a所示。
精密称取26mg干燥的石榴皮粉末分别置于研钵中,并称取32.5mg碳分子筛加入研钵中与石榴皮粉末混合研磨90s。取1ml规格固相萃取小柱,底部均加入筛板,分别用研磨好的固体混合物填充小柱,顶部均加入筛板,压实后置于固相萃取仪上。用0.2ml的甲醇对小柱进行洗脱,底部用1.5ml规格离心管收集洗脱液。最后,将洗脱液在离心仪中以13000rpm离心5分钟,取上层清夜,用uhplc-q-tof/ms进行分析。
石榴皮基质固相微萃取提取液的uhplc-q-tof/ms总离子流图和提取离子流图如图6b所示。
表2.5种成分的标准曲线以及检测限和定量限汇总表
回收率实验
精密称取26mg干燥的石榴皮粉末分别置于研钵中,并称取32.5mg碳分子筛加入研钵中与石榴皮粉末混合研磨90s。取1ml规格固相萃取小柱,底部均加入筛板,分别用研磨好的固体混合物填充小柱,顶部均加入筛板,压实后置于固相萃取仪上。用0.2ml的甲醇对小柱进行洗脱,底部用1.5ml规格离心管收集洗脱液。最后,将洗脱液在离心仪中以13000rpm离心5分钟,取上层清夜,用uhplc-q-tof/ms进行分析。平行做3组,其中一组加混合对照品至浓度为2μg/ml,另一组加混合对照品至浓度为20μg/ml,剩余一组不加混合对照品。
表3重复性、含量测定、回收率实验结果汇总表
根据表中数据显示,本发明方法灵敏度高,重复性良好,回收率高,具有较高的检测准确度。
- 还没有人留言评论。精彩留言会获得点赞!