小儿扶脾颗粒的标准指纹图谱的构建方法及其应用与流程
本发明涉及一种小儿扶脾颗粒的标准指纹图谱的构建方法及其应用,属于中药的质量控制领域。
背景技术:
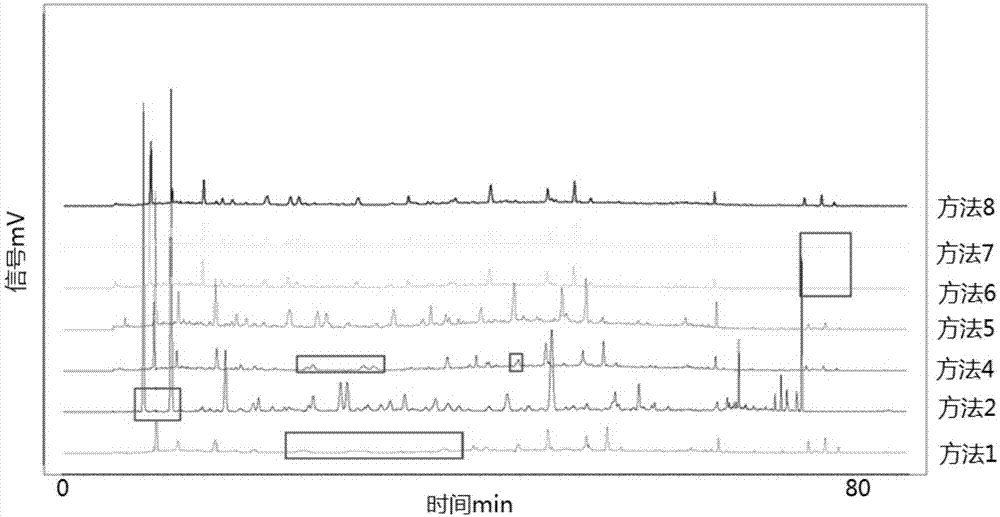
小儿扶脾颗粒由白术、陈皮、山楂、党参、莲子、茯苓6味中药制成,具有健脾胃、助消化的功效,用于小儿脾胃气血,消化不良,体质消瘦。中药指纹图谱是指采用一定分析手段得到能够标定中药特性的共有峰图谱,能够综合反映该制剂的主要组分的情况,是一种综合的、宏观的、整体的、可量化的鉴别手段。目前,利用中药指纹图谱评价中药产品的批次一致性已经被国内外广泛接受和认可。现有技术中尚无有关小儿扶脾颗粒的质量控制方法的明确报道,因此,为了更加全面有效的控制小儿扶脾颗粒的质量,保证其疗效,提供一种小儿扶脾颗粒的质量控制方法更全面有效的控制其质量具有重大意义。技术实现要素:本发明解决的技术问题是,针对现有技术的不足,构建一种新的小儿扶脾颗粒的标准指纹图谱。基于该标准指纹图谱,进一步提供一种小儿扶脾颗粒指纹图谱检测方法,该方法能够更全面有效的控制小儿扶脾颗粒的质量。本发明的技术方案是,提供一种小儿扶脾颗粒的标准指纹图谱的构建方法,分别将不同生产批次的小儿扶脾颗粒样品制成溶液a,采用高效液相色谱法,得到若干小儿扶脾颗粒的指纹图谱;确定小儿扶脾颗粒的指纹图谱的共有峰的峰位置及其峰面积,并将这些共有峰的峰位置和峰面积作为小儿扶脾颗粒的标准指纹图谱;高效液相色谱的条件:色谱柱为十八烷基硅烷键合硅胶柱;以乙腈为流动相a,以体积分数0.1%的磷酸水溶液为流动相b,梯度洗脱,流速为0.8-1.2ml/min;检测波长为220-360nm;柱温25-35℃。在高效液相色谱中,共有峰的峰位置是通过保留时间确定的。通过测定若干批次样品的指纹图谱,得到共有峰的峰位置(相对)和峰面积(相对)分别如表1和表2所示。其中以第10号峰-橙皮苷作为参照,计算相对保留时间和相对峰面积。优选地,小儿扶脾颗粒的标准指纹图谱为:表1共有峰的峰位置峰号相对保留时间10.16-0.1820.27-0.2830.40-0.4140.44-0.4650.46-0.4760.58-0.5970.67-0.6980.83-0.8590.94-0.95101111.26-1.28121.47-1.49表2共有峰的峰面积峰号相对峰面积10.18-2.1520.14-0.9130.32-0.7740.18-0.5450.17-0.6060.17-0.7270.20-0.5080.27-1.1890.41-0.77101110.17-0.38120.05-0.50。优选地,梯度洗脱为:0-100min,5%a-95%a。优选地,色谱柱:ulimatexb-c18,柱长为250mm,内径为4.6mm,粒径为3.5μm。优选地,流速为1.0ml/min;柱温30℃。优选地,溶液a的制备方法为:称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,精密加入25ml、体积分数为50%的甲醇水溶液,称定重量,摇匀后超声提取30分钟,放冷,再称定重量,体积分数为50%的甲醇水溶液补足减失的重量,摇匀,3000rpm离心10分钟,离心后的上清液用0.22μm过滤头滤过,即得。优选地,超声提取的工艺参数为:超声时间30min,频率40khz;离心的工艺参数为:转速3000rpm,时间10min。优选地,梯度洗脱程序为:表3梯度洗脱程序时间(min)0.1%磷酸(%)乙腈(%)0.019555901025871355703080307088109089955100955上表中,0.01min-5min,流动相由95%的0.1%磷酸-5%的乙腈逐步变化为90%的0.1%磷酸-10%的乙腈。优选地,小儿扶脾颗粒的处方为:白术47.6g、陈皮23.8g、山楂47.6g、党参47.6g、莲子47.6g、茯苓38g、蜂蜜(炼)47.6g、蔗糖950g;制法为:以上六味药材,加水煎煮二次,第一次1.5小时,第二次2小时,合并煎液,滤过,滤液浓缩至相对密度为1.10(60℃)的清膏,加炼蜜,混匀,再加蔗糖,制成颗粒,干燥,即得。本发明还提供上述标准指纹图谱在小儿扶脾颗粒样品检测中的应用。小儿扶脾颗粒样品检测的具体方法为:(1)按上述方法获得标准指纹图谱,即:分别将不同生产批次的小儿扶脾颗粒样品制成溶液a,采用高效液相色谱法,得到若干小儿扶脾颗粒的指纹图谱;确定小儿扶脾颗粒的指纹图谱的共有峰的峰位置及其峰面积,并将这些共有峰的峰位置和峰面积作为小儿扶脾颗粒的标准指纹图谱;高效液相色谱的条件:色谱柱为十八烷基硅烷键合硅胶柱;以乙腈为流动相a,以0.1%磷酸为流动相b,梯度洗脱,流速为0.8-1.2ml/min;检测波长为220-360nm;柱温25-35℃;(2)按与步骤(1)中溶液a相同的制备方法将待测小儿扶脾颗粒样品制成溶液b,采用高效液相色谱法,并使高效液相色谱的条件与步骤(1)的高效液相色谱的条件相同,得到待测小儿扶脾颗粒样品的指纹图谱;(3)将待测小儿扶脾颗粒样品的指纹图谱与小儿扶脾颗粒的标准指纹图谱进行比对,根据指纹图谱的相似度确定待测小儿扶脾颗粒样品的质量。相似度越高,说明待测小儿扶脾颗粒样品质量越好。本发明的有益效果是,该方法通过建立指纹图谱获得不同批次间小儿扶脾颗粒12个共有的色谱峰,构建了小儿扶脾颗粒标准指纹图谱,基于小儿扶脾颗粒标准指纹图谱,可以得到一种新的小儿扶脾颗粒质量检测方法,用于更好地评价和控制小儿扶脾颗粒的质量。附图说明图1为供试品溶液的制备考察图谱。图2为洗脱程序的考察对比图谱。图3a和图3b均为波长的考察对比图谱。图4为色谱柱选择图谱。图5为柱温考察图谱。图6为流速考察图谱。图7为精密度考察图谱。图8为稳定性考察图谱。图9为重复性考察图谱。图10为对照指纹图谱。具体实施方式下面结合实施例对本发明作进一步说明。实施例1:小儿扶脾颗粒指纹图谱优选试验。小儿扶脾颗粒由白术、陈皮、山楂、党参、莲子、茯苓6味中药制成,具有健脾胃、助消化的功效,用于小儿脾胃气血,消化不良,体质消瘦。本实验针对其处方中药进行指纹图谱研究,可从整体特征把握中药的品种和质量情况。仪器与试药lc-20at型高效液相色谱仪(日本岛津公司);分析天平(梅特勒-托利多仪器(上海)有限公司);恒温水浴锅(江苏金坛市中大仪器厂);电子调温电热套(天津泰斯特仪器有限公司);离心机(江苏金坛市中大仪器厂);移液器(百得实验室仪器(苏州)有限公司)超声波清洗器(sb-5200d,宁波新芝生物科技股份有限公司);旋转蒸发仪(上海亚荣生化仪)。橙皮苷对照品(由中国食品药品检定研究院提供);小儿扶脾颗粒12批均由湖南时代阳光医药有限公司提供(批号同含量测定)。乙腈为色谱纯;水为重蒸水;其余试剂均为分析纯。药品来源:本研究所用小儿扶脾颗粒由湖南时代阳光药业股份有限公司生产,批号:20160901、20160505、20160708、20161206、20170206、20170901、20180102、20180103、20180104、20180105、20180106、20180107。通过与对照品比对,小儿扶脾颗粒指纹图谱中主要已知化合物为橙皮苷,含量较高,易获得,故选择橙皮苷为本品指纹图谱参照物。提取方法的考察方法1:称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,加入25ml的甲醇,摇匀后于70℃水浴锅中水浴30分钟后取出,自然冷却后3000rpm离心10分钟,离心后的上清液用0.22μm过滤头滤过,取续滤液,即得。方法2:称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,加入25ml水,摇匀后于70℃水浴锅中水浴30分钟后取出,自然冷却后3000rpm离心10分钟,离心后的上清液用50ml乙酸乙酯萃取一次,乙酸乙酯层经旋转蒸发蒸干,用水溶解蒸干物并定容至5ml,滤过,取续滤液,即得。方法3:称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,加入25ml水,摇匀后于70℃水浴锅中水浴30分钟后取出,自然冷却后3000rpm离心10分钟,离心后的上清液用50ml石油醚萃取一次,石油醚层经旋转蒸发蒸干,用水溶解蒸干物并定容至5ml,滤过,取续滤液,即得。方法4:称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,加入12.5ml的水和12.5ml的甲醇,摇匀后于70℃水浴锅中水浴30分钟后取出,自然冷却后3000rpm离心10分钟,离心后的上清液用0.22μm过滤头滤过,取续滤液,即得。方法5:称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,加入15ml的水和15ml的甲醇,摇匀后于70℃水浴锅中水浴30分钟后取出,自然冷却后3000rpm离心10分钟,离心后的25ml上清液置于蒸发皿中,水浴蒸至无醇味,用水溶解,转移至10ml量瓶中,定容至刻度。用0.22μm过滤头滤过,取续滤液,即得。方法6:称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,加入25ml的水,摇匀后于100℃水浴锅中水浴回流30分钟后取出,自然冷却后3000rpm离心10分钟,离心后的上清液用0.22μm过滤头滤过,取续滤液,即得。方法7:称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,加入25ml的水,摇匀后超声提取30分钟取出,3000rpm离心10分钟,离心后的上清液用0.22μm过滤头滤过,取续滤液,即得。方法8:称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,精密加入25ml50%甲醇-水,称定重量,摇匀后超声提取30分钟,放冷,再称定重量,用50%甲醇-水补足减失的重量,摇匀,3000rpm离心10分钟,离心后的上清液用0.22μm过滤头滤过,取续滤液,即得。见附图1(附图1的方框表示各个方法中的特点或为不选取的理由)和表4:表4供试品溶液制备方法及其考察结果结果表明,50%甲醇超声提取效果最佳,因此本品的供试品溶液的制备方法选择方法8,即称取4.0g小儿扶脾颗粒,置于100ml锥形瓶中,精密加入25ml50%甲醇-水,称定重量,摇匀后超声提取30分钟,放冷,再称定重量,用50%甲醇-水补足减失的重量,摇匀,3000rpm离心10分钟,离心后的上清液用0.22μm过滤头滤过,即得。实施例2:色谱条件的考察流动相的选择:色谱柱固定为ulimatexb-c18,比较了甲醇-水,不同比例的等度洗脱,乙腈-0.1%磷酸,不同比例的等度洗脱及不同梯度洗脱系统。结果表明②乙腈-0.1%磷酸,梯度洗脱,对小儿扶脾颗粒的各种成分有很好的分离效果,能提供丰富的色谱信息。甲醇-水系统的热力学和可压缩性因素,在梯度洗脱时易导致基线漂移;且甲醇-水系统柱压相对较高,而乙腈-水相对较好。因此确定乙腈-0.1%磷酸为流动相。梯度洗脱程序的选择:将色谱柱固定为ulimatexb-c18,考察不同洗脱程序(见下表),结果以洗脱程序ⅲ所得的色谱图中各组分的峰清晰可辨识,分离度大于1.5,峰形尖锐,保留时间适中,基线漂移较弱,指纹图谱包含的信息丰富,见附图2。表5洗脱程序ⅰ表6洗脱程序ⅱ表7洗脱程序ⅲ实施例3:波长的选择采用紫外检测器对检测波长进行考察,比较了220nm,240nm,260nm,280nm,300nm,310nm,320nm,330nm,340nm,360nm条件下的色谱图。经过比较发现,已知小儿扶脾颗粒标志成分橙皮苷的最大吸收波长为283nm,但是280nm并非指纹图谱的最佳波长,因为在此条件下橙皮苷的峰高太高,峰面积太大,其他峰不明显,无法辨认,谱图中的各峰的峰高不协调。检测波长为330nm时,谱图中各峰的峰高协调,所有的峰都可辨认,此时的谱图呈现的信息最丰富,因此确定检测波长为330nm,见附图3a和图3b。实施例4:色谱柱的选择洗脱程序固定为洗脱程序ⅲ,对不同品牌的色谱柱进行考察,分别为色谱柱1:ulimatexb-c18(4.6*250mm,5um)序列号211603287;色谱柱2:agilentzorbaxsb-c18(4.6*250mm,5um)序列号uscl041432见附图4。实施例5:柱温的选择洗脱程序固定为洗脱程序ⅲ,色谱柱固定为色谱柱1,分别对柱温25℃、30℃、35℃进行考察,结果显示档当柱温为30℃时所得的指纹图谱各色谱峰分离效果较好。见附图5。实施例6:流速的选择洗脱程序固定为洗脱程序ⅲ,色谱柱固定为色谱柱1,柱温固定为30℃,分别对流速0.8ml/min、1.0ml/min、1.2ml/min所得图谱进行比较,结果显示流速为1.0ml/min时所得指纹图谱各色谱峰分离效果较好。见附图6。综上所述,小儿扶脾颗粒指纹图谱检测方法最佳条件为:以ulimatexb-c18为色谱柱;柱温箱温度为30℃;进样液体积:10μl;检测波长:330nm;流动相总流速:1.0ml/min。实施例7:指纹图谱方法学考察1.精密度试验精密称取同一小儿扶脾颗粒(批号:20180102)4.0g,按实施例1的方法8制备供试品溶液,连续进样6次进行测定,测定结果见表8至表10各共有峰与内参比峰(橙皮苷-即第10号峰)相对保留时间和相对峰面积的rsd均小于5%,结果相似度大于0.99,见表10,表明精密度良好,见附图7(图7-图9中右侧的s1(12)中的s1代表第一条谱线,(12)代表12个峰)。小儿扶脾颗粒指纹图谱精密度考察结果如下(表8-10)。表8精密度考察结果12个共有峰的相对保留时间表9精密度考察结果12个共有峰的相对峰面积表10小儿扶脾颗粒指纹图谱精密度测定结果(相似度)2.稳定性试验精密称取同一小儿扶脾颗粒(批号:20180102)4.0g,按方法8制备供试品溶液,连续进样6次进行测定,测定结果见表11至表13,,各共有峰与内参比峰(橙皮苷)相对保留时间和相对峰面积的rsd均小于5%,结果相似度大于0.98,见表13,表明精密度良好,见附图8。小儿扶脾颗粒指纹图谱稳定性考察结果如下(表11-13)。表11稳定性考察结果12个共有峰的相对保留时间表12稳定性考察结果12个共有峰的相对峰面积表13小儿扶脾颗粒指纹图谱稳定性测定结果(相似度)3.重复性试验精密称取同一小儿扶脾颗粒(批号:20180102)4.0g,按“5.1”项下的方法8制备供试品溶液,连续进样6次进行测定,测定结果见表14和表16,,各共有峰与内参比峰(橙皮苷)相对保留时间和相对峰面积的rsd均小于5%,结果相似度大于0.99,见表,16,表明精密度良好,见附图9。小儿扶脾颗粒指纹图谱重复性考察结果如下:(表14-16)表14重复性考察结果12个共有峰的相对峰面积表15重复性考察结果12个共有峰的相对保留时间表16小儿扶脾颗粒指纹图谱精密度测定结果(相似度)根据以上方法学考察结果,表明该方法测定小儿扶脾颗粒的指纹图谱,精密度、重复性、稳定性均较好,能够准确测定该制剂的指纹图谱。实施例8:小儿扶脾颗粒指纹图谱检测、标准指纹图谱的获得及相似度的确定收集13批小儿扶脾颗粒样品,按上述供试品溶液的制备方法制备供试品溶液,依法测定,计算各共有峰的相对保留时间,相对峰面积和相似度,用相似度软件以这13批样品指纹图谱为基础获得“共有模式”作为标准指纹图谱,见附图10,结果见表16-17。表16共有峰的相对保留时间表16共有峰的相对保留时间峰号20180104201801052018010620180107201801082018010410.170.170.170.170.160.1720.280.270.270.270.260.2830.400.400.400.400.380.4040.450.450.450.450.430.4550.470.460.460.460.440.4760.580.580.580.580.560.5870.680.690.680.680.810.6880.840.830.840.840.920.8490.950.950.950.950.970.9510(s)1.001.001.001.001.001.00111.271.271.271.271.241.27121.481.481.481.481.441.48表17小儿扶脾颗粒指纹图谱13批供试品测定结果(相似度)13批小儿扶脾颗粒指纹图谱与标准指纹图谱计算相似度,其结果均大于0.85,确定小儿扶脾颗粒指纹图谱与对照指纹图谱经中药色谱指纹图谱相似度评价系统计算,相似度不得低于0.85。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1