细胞自食的增强剂用于使肿瘤对放射疗法敏感的用途的制作方法
概述
本发明涉及表1和表2中突出显示的化合物及其类似物在癌细胞中用于增强ir-介导的细胞自食(cannibalism)的用途。所述化合物在本文中称为“ir介导的细胞自食的增强剂”。它们可用于在将接受或已接受放射疗法治疗的受试者中增强肿瘤免疫原性和/或诱导显著的保护性抗癌免疫应答。换句话说,所述化合物可用于有此需求的受试者中加强放射疗法治疗。所述化合物优选选自:美海屈林1,5-萘二磺酸盐、氟比洛芬、米那普利二盐酸盐、杨梅素、地高辛、洋地黄毒甙、毛花洋地黄甙、lopa87、vp331、rn-1-026、sg6163f、vp450和vp43。
背景技术:
放射疗法是癌症治疗中最常用的抗肿瘤策略之一。超过一半的癌症患者接受了放射疗法治疗。使用放射疗法(单独或与手术和化学疗法联合)对于治疗头颈部、乳腺、肺、前列腺、消化道的肿瘤和宫颈癌至关重要。肿瘤组织暴露于电离辐射(ir)引发各种致死过程,例如细胞凋亡、自噬性细胞死亡、有丝分裂障碍(catastrophe)或衰老。
这些机制有助于在经放射的组织中破坏癌细胞,但也可以诱导经放射的肿瘤细胞的免疫原性2,3,改变肿瘤微环境并允许产生抗肿瘤免疫应答,包括危险信号(三磷酸腺苷和hmgb1蛋白)的释放4,5、嘌呤能受体(特别是p2x7和p2y2)4、tlr4受体2,3和nlrp3炎性体4的激活。通过描述对非放射的癌细胞的远程效应,来说明局部肿瘤反应和免疫系统刺激之间的关系。这种奇异效应,也称为异位(abscopal)效应,已在不同的小鼠肿瘤模型以及患者中进行了多年的研究6-9。其检测与存在大量促炎细胞因子(如tnfα)、产生干扰素(ifn)γ的t细胞呈正相关,需要产生可待观察到的有效免疫应答10,11。这种效果也可以在用放射疗法联合免疫调节剂(如伊匹单抗(ipilimumab)或白细胞介素-2)治疗的患者中观察到12。对异位效应的描述强化了最近在许多动物模型中的描述,即免疫系统各组分对放射疗法应答的主要贡献。通过免疫检查点调节剂的治疗调节(例如使用针对ctla4或pd1的单克隆抗体)或巨噬细胞激活(使用针对csf1r的单克隆抗体)的影响,最近已研究并强调了免疫细胞(包括肿瘤相关巨噬细胞、t淋巴细胞和树突细胞)的核心作用。尽管放射疗法在抗肿瘤治疗库中具有核心作用,但参与消除肿瘤细胞的生物学过程仍然未充分定义且有争议。关于放射诱导的肿瘤细胞死亡的早期研究相对较老,并且其先于最近发现新的细胞死亡过程以及免疫系统在放射后消除肿瘤细胞中的核心作用。
近年来科学和技术的进步揭示了细胞死亡的几个过程的存在。已经提出了主要基于形态学和功能标准的细胞死亡模式的分类13,并将致死过程细分为三种不同的类型:i型细胞死亡(也称为细胞凋亡)、ii型细胞死亡(或自噬)和iii型细胞死亡(或坏死)。最初的组织构架是建立在i型和iii型细胞死亡模式之间的明显区别之上,建立在受调节或意外诱导的过程之间,或通过区分诱导体内旁路炎症反应相关的细胞死亡,与被认为沉默的或致耐受性的致死过程,这种分类仍然将ii型细胞死亡误称为自噬(其主要是维持细胞内稳态所需的细胞内存活机制)并未反映出坏死性死亡受到遗传控制或具有免疫原性的倾向,正如最近坏死性细胞凋亡的分子特征所揭示的14,15。此外,细胞死亡的子程序(subroutine)没有或部分揭示这些传统的形态、代谢和生物化学修饰(如有丝分裂死亡和角质化),关于细胞死亡子程序的研究较少,并被分组在定义不明的细胞死亡模式亚组中,也称为非典型细胞死亡亚组13。
近年来,已经描述了几种新的细胞死亡机制(例如细胞侵入性死亡(entosis)或叠套凋亡(emperitosis)),并且与这种被忽略的细胞死亡模式亚组相关16,17。这些非典型死亡模式的特征突出了细胞死亡过程的存在,而这种过程并不像典型的细胞死亡模式那样以细胞自主方式实现,而是在邻近细胞吞噬活细胞后引发。
几十年来,非细胞自主死亡(ncad)已经被偶然观察和研究。最初称为细胞吞噬18,19或者细胞侵入现象(emperipolesis)20,这些过程参与对红细胞、中性粒细胞、血小板或t细胞稳态的控制18,21,22,因此被认为是体内细胞死亡的主要生理形式18。在原代细胞的离体培养过程中23或在生理过程的组织学分析(例如胚胎植入24和自身反应性t细胞的肝内消耗25)或人类疾病(包括癌症23、炎症综合征23、在感染性疾病期间26,27)期间也观察到ncad,其被检测为细胞-内-细胞(cell-in-cell)结构。主要在人类肿瘤(包括乳腺、宫颈以及结肠癌或黑素瘤)中的恶性细胞之间的同型相互作用后检测到ncad,在肿瘤细胞和基质细胞28之间、肿瘤细胞和免疫效应细胞(如淋巴细胞29和nk细胞30)之间的异型相互作用后,也检测到ncad,但也可以在免疫效应细胞和上皮细胞之间的相互作用后观察到(通过胸腺护理细胞中t细胞的破坏31或肝脏中自身反应性t细胞25所揭示的)。最近,ncad也与传染病有关。在体外共培养期间,检测到未感染细胞吞噬人类免疫缺陷病毒1(hiv-1)或埃巴病毒(ebv)感染的细胞,并且降解内化的感染细胞,这被提议为感染细胞和宿主细胞之间进行病毒传播的新细胞-至-细胞(cell-to-cell)模式的第一步26,27,强调了ncad也有助于微生物发病机制的能力。
非细胞自主死亡程序的第一步开始于两个细胞伙伴通过膜粘附受体(如e-或p-钙粘蛋白)或应激受体(如脂蛋白受体相关蛋白)的相互作用,然后形成相互作用细胞之间的粘附连接,其激活两个相互作用细胞上的信号传导途径,并且可能涉及小gtp酶(例如rho32和cdc4233)和rock激酶16。然后,显示肌动球蛋白收缩性的调节和肌动蛋白细胞骨架在“靶”细胞水平上的重新组织有利于它们侵入宿主细胞32,34。这个过程不同于细胞自食,所述细胞自食也可以通过激活宿主细胞上的特定信号传导通路(如吞噬作用相关的信号通路,包括细胞骨架重塑细胞分裂周期42(cdc42)、趋化因子(c-x-c基序)配体1(cxcl1)或趋化因子(c-x-c基序)配体6(cxcl6))而触发ncad,并导致靶细胞的活性吞噬33。一旦内化,被吞噬的细胞可被“宿主”溶酶体酶(例如组织蛋白酶和颗粒酶)靶向,并通过可能涉及几种典型细胞死亡调节剂(例如细胞色素c、半胱天冬酶或自噬相关(atg)蛋白)的不同致死机制被消除。细胞侵入性死亡(一种非细胞自主死亡)最初在乳癌细胞之间的同型相互作用之后被描述,是细胞-内-细胞侵袭机制,其不需要激活半胱天冬酶来消除被吞噬的细胞16。相反,肿瘤细胞对自然杀伤(nk)的吞噬引发了叠套凋亡(一种需要半胱天冬酶3激活和dna片段化的程序性细胞-内-细胞死亡30),这揭示了可以通过半胱天冬酶依赖性或半胱天冬酶非依赖性过程来消除被吞噬的细胞。
尽管付出了相当大的努力,但在过去10年中为增强放射疗法的有效性而开发的方法尚未达到预期的临床成功。在先前方法失败的原因中,可以指出迄今为止开发的大多数方法具有与当前项目不同的特征。此外,使用合理方法尚未开发出基于细胞死亡和治疗耐受性机制表征的这些方法。具体而言,他们从未考虑过免疫系统对肿瘤细胞消除的影响。最后,他们仅转换来自医学肿瘤学领域的放射疗法应用,而没有考虑放射后细胞死亡的具体机制。
在这种情况下,需要鉴定和表征放射疗法照射后细胞死亡的调节剂是什么,并评估在癌症患者中施用这些调节剂的免疫学后果是什么。
技术实现要素:
尽管实施了密集的生物和药物研究以更好地表征与抗癌治疗相关的细胞和生物化学过程,但是作为临床中最常用的抗癌治疗之一的放射疗法,其治疗效果的致死机制仍然是未知的。已经响应于电离辐射而检测到的致死过程(例如细胞凋亡和有丝分裂障碍)并不直接涉及治疗效果35,这表明仍然未知的其他细胞死亡模式可能有助于放射疗法的治疗效果。
本项目基于发现新的细胞死亡机制(细胞自食),这种机制在ri或其他抗癌治疗后从未被其他人检测到。通常,在肿瘤疫苗接种实验中,癌细胞在体外暴露于细胞毒性抗癌治疗,直到70%的细胞在质膜外部小叶上暴露磷脂酰丝氨酸48。有趣的是,目前的结果表明,基于其增强ir-介导的细胞自食(ircce)的能力而选择的化合物,将癌细胞转化为刺激抗肿瘤免疫应答的疫苗。更具体地说,本结果首次揭示了,在没有显著增加经处理和经放射的细胞死亡的情况下,ircce+ir的组合引发ir介导的抗癌免疫应答(图5a),表明细胞自食或细胞自食相关的信号传导通路有助于在放射后诱导肿瘤免疫原性。
放射通常用于治疗癌症。通常与化学疗法和/或手术组合,放射疗法包括局部和全身施用以及许多新进展,包括放射免疫疗法。放射对赘生细胞的细胞毒性作用源于放射在细胞内dna分子的一条或两条链中引起断裂的能力。细胞周期所有阶段的细胞都易受这种影响。然而,dna损伤在癌细胞中更可能是致死性的,因为它们不太能够修复dna损伤。具有功能性细胞周期检查点蛋白和修复酶的健康细胞更有可能在治疗后修复放射损伤并正常发挥功能。然而,放射会带来副作用,这对患者来说是一种负担。放射的副作用类似于化疗法的副作用,并且由于同样的原因产生,即健康组织的损伤。放射通常比化疗更局部化,但是这种治疗仍然伴随着对先前健康组织的损害。许多副作用都令人不愉快,而放射与化学疗法其本身都具有致突变性、致癌性和致畸性的缺点。虽然正常细胞通常在治疗后两小时内开始从治疗中恢复,但可以在健康细胞的基因中诱导突变。这些风险在某些组织中升高,例如生殖系统中的组织。而且,已经发现不同的人放射耐受不同。在一个个体中可能不会导致新癌症的剂量实际上可能在另一个个体中产生其他癌症。这可能是由于细胞周期检查点蛋白或修复酶中预先存在的突变,但目前的做法无法预测特定个体存在风险的剂量。放射的常见副作用包括膀胱刺激、疲劳、腹泻、低血细胞计数、口腔刺激、味觉改变、食欲不振、脱发、皮肤刺激、肺功能改变、肠炎、睡眠障碍等。
为患者提供与放射发挥协同作用的加强剂是非常有利的,从而使患者较少暴露于放射疗法。这确实会减少上述副作用,同时实现改善的有益效果。这也是本发明的一个方面。
总之,有人提出,鉴于细胞自食对消除经放射的癌细胞的突出重要性以及在肿瘤对放射反应中免疫应答的重要性日益增加,在癌症治疗期间细胞自食是应该受到青睐的“理想”死亡,从而有效消除经放射的癌细胞。这可以通过向患者施用细胞自食增强剂来实现,如本发明人在下面的实验部分中所证明的。
定义
术语“放射疗法”在本领域中通常用于指多种类型的放射疗法,包括内部和外部放射疗法、放射免疫疗法、以及使用各种类型的放射,包括x射线、γ射线、α粒子、β粒子、光子、电子、中子、放射性同位素和其他形式的电离辐射。除非另有说明,否则本文所用的术语“辐射疗法”、“放射疗法”、“放射”和“辐射”包括所有这些类型的放射疗法。有不同类型的放射疗法机器,它们的工作方式略有不同。放射疗法疗程的次数和持续时间取决于癌症的类型及其在体内的位置。表浅皮肤癌可能只需要很少的短期治疗,而体内较深的癌症可能需要更长时间的治疗。
术语“抑制肿瘤生长”、“治疗肿瘤生长”和“治疗癌症”等是指在用本公开的组合物、试剂盒或方法治疗后,降低肿瘤生长速度、完全停止肿瘤生长、导致现有肿瘤大小消退、根除现有肿瘤和/或预防其他肿瘤的发生。“抑制”肿瘤细胞生长意指任何或所有以下状态:减缓、延迟和停止肿瘤生长以及肿瘤缩小。肿瘤的“延迟进展”意指推迟、阻碍、减缓、延缓、稳定和/或延迟疾病的进展。该延迟可以具有不同的时间长度,这取决于病史和/或被治疗的个体。可以通过本领域已知的任何方法评估肿瘤细胞生长,包括但不限于测量肿瘤大小、使用3h-胸苷掺入分析确定肿瘤细胞是否增殖、或计数肿瘤细胞。
如本文所用,当涉及本公开化合物与放射联合的组合施用时,“协同”或“协同作用”是指与单独施用化合物和放射相比,该组合作用大于二者的加和。
在本申请的上下文中,“加强”是指增强或增加例如放射疗法治疗的效果、或促进或加强例如生化或生理作用或效果。
“有效量”是指如本文所述的化合物的量,其可以治疗上有效地治疗与本公开相关的癌症。这些化合物所需的精确量将随所用的具体化合物或衍生物、待治疗的受试者年龄和状况、以及病症的性质和严重程度而变化。然而,可由本领域普通技术人员仅通过常规实验确定有效量。本领域普通技术人员无需过度实验即可确定有效量的放射。放射参数(例如给药量和频率)在本领域中是公知的。
“药学上可接受的”是指在合理医学判断范围内,匹配合理的利益/风险比的适合与人类和动物组织接触而没有过度毒性、刺激、过敏反应、或其他问题并发症的那些化合物、材料、组合物和/或剂型。
“药学上可接受的盐”是指所公开化合物的衍生物,其中母体化合物通过制备其酸或碱盐来修饰。本公开的化合物与各种有机和无机酸和碱形成酸和碱加成盐,并包括通常用于药物化学的生理学上可接受的盐。这些盐也是本公开的一部分。用于形成这种盐的典型无机酸包括盐酸、氢溴酸、氢碘酸、硝酸、硫酸、磷酸、连二磷酸等。还可以使用衍生自有机酸的盐,例如脂肪族单羧酸和二羧酸、苯基取代的烷酸、羟基烷酸和羟基烷二酸、芳族酸、脂肪族和芳族磺酸。因此,这样的药学上可接受的盐包括乙酸盐、苯基乙酸盐、三氟乙酸盐、丙烯酸盐、抗坏血酸盐、苯甲酸盐、氯苯甲酸盐、二硝基苯甲酸盐、羟基苯甲酸盐、甲氧基苯甲酸盐、甲基苯甲酸盐、邻乙酰氧基苯甲酸盐、萘-2-苯甲酸盐、溴化物、异丁酸盐、苯基丁酸盐、β-羟基丁酸盐、丁炔-1,4-二酸盐、己炔-1,4-二酸盐、cabrate、辛酸盐、氯化物、肉桂酸盐、柠檬酸盐、甲酸盐、富马酸盐、乙醇酸盐、庚酸盐、马尿酸盐、乳酸盐、苹果酸盐、马来酸盐、羟基马来酸盐、丙二酸盐、扁桃酸盐、甲磺酸盐、烟酸盐、异烟酸盐、硝酸盐、草酸盐、邻苯二甲酸盐、对苯二甲酸盐、磷酸盐、磷酸氢盐、磷酸二氢盐、偏磷酸盐、焦磷酸盐、丙炔酸盐、丙酸盐、苯丙酸盐、水杨酸盐、癸二酸盐、琥珀酸盐、辛二酸盐、硫酸盐、硫酸氢盐、焦硫酸盐、亚硫酸盐、重亚硫酸盐、磺酸盐、苯磺酸盐、对溴苯磺酸盐、氯苯磺酸盐、乙磺酸盐、2-羟基乙磺酸盐、甲磺酸盐、萘-1-磺酸盐、萘-2-磺酸盐、对甲苯磺酸盐、二甲苯磺酸盐、酒石酸盐等。通常用于形成盐的碱包括氢氧化铵和碱和碱土金属氢氧化物、碳酸盐、以及脂肪族和伯、仲和叔胺、脂肪族二胺。特别适用于制备加成盐的碱包括氢氧化钠、氢氧化钾、氢氧化铵、碳酸钾、甲胺、二乙胺和乙二胺。
替代实施方案提供了如上所述的加强放射疗法癌症治疗方法,包括施用“缓释制剂”,其能够以受控的、非即时方式、时间依赖的方式将活性成分释放到周围环境中。该缓释制剂可包含可生物降解的聚合物,例如选自乳酸的同聚物;乙醇酸的同聚物;聚-d,l-乳酸和乙醇酸的共聚物;黄体激素释放激素(lhrh)类似物的水不溶性肽盐;聚(磷酸酯);双(对羧基苯氧基)丙烷(cpp)与癸二酸共聚物;聚酸酐聚合物;聚(丙交酯)-共-乙交酯)聚乙二醇共聚物;和乙烯-乙酸乙烯酯共聚物。
在该方法的一些具体实施方案中,缓释制剂在四周或更长时间内,或者在一周或更长时间内,或者在几小时或更长时间内释放ircce药物。
本文所述的方法、组合物制剂和用途适用于人和动物,优选哺乳动物。
因此,如本文所用,术语“受试者”涉及任何哺乳动物,包括小鼠、大鼠、猪、猴和马。在具体的实施方案中,它指人。在本发明的上下文中,“有此需求的受试者”或“患者”旨在包括将受益于或可能受益于本发明的方法和药物组合物的任何受试者。受试者可能患有任何阶段的癌症,从而它可能是早期非侵入性癌症,或者可能是已经发展成在体内转移的晚期癌症。
“患者”在本文中是指动物,包括哺乳动物,优选人。
如本文所用,术语“癌症”旨在包括任何形式的癌症或肿瘤。癌症的非限制性实例包括脑癌(例如,神经胶质瘤)、胃癌、头颈癌、胰腺癌、非小细胞肺癌、小细胞肺癌、前列腺癌、结肠癌、非霍奇金淋巴瘤、肉瘤、睾丸癌、急性非淋巴细胞白血病和乳癌。在具体的实施方案中,脑癌是星形细胞瘤,更具体地,是多形性胶质母细胞瘤;肺癌是小细胞肺癌或非小细胞肺癌;以及,头颈癌是鳞状细胞癌或腺癌。
本发明基于以下发现:已知和未知的药物能够在携带肿瘤的动物体外和体内增强ir介导的细胞自食。
如本文所用,术语“细胞自食诱导药物”、“细胞自食诱导剂”、“细胞自食诱导化合物”和“细胞自食增强剂”表示能够增强放射触发细胞自食的能力的化合物。在这些术语下,指定能够增强ir-介导的细胞自食(以下称为ircce)的化合物。所选细胞自食调节剂的化学结构列于表1和表2中。
表1
表2
总的来说,本发明人发现了16种有效的细胞自食增强剂化合物,即:8-氮鸟嘌呤、美海屈林1,5-萘二磺酸盐、氟比洛芬、米那普利二盐酸盐、杨梅素、地高辛、洋地黄毒甙、毛花洋地黄甙、盐酸多柔比星、lopa87、vp331、rn-1-026、sg6163f、vp450、vp43。还发现了许多有趣的化合物。它们例如是lopa87的类似物(lopa90、lopa93、lopa94、lopa101、lopa104、lopa105、lopa106)和sg6143f的类似物(sg6144、sg6146和sg6149)。因此,在优选的实施方案中,本发明的细胞自食增强剂选自:8-氮鸟嘌呤、美海屈林1,5-萘二磺酸盐、氟比洛芬、米那普利二盐酸盐、杨梅素、地高辛、洋地黄毒甙、毛花洋地黄甙、盐酸多柔比星、lopa87、vp331、rn-1-026、sg6163f、vp450和vp43,其结构见表1和表2所示。在更优选的实施方案中,本发明的细胞自食增强剂选自:美海屈林1,5-萘二磺酸盐、氟比洛芬、米那普利二盐酸盐、杨梅素、地高辛、洋地黄毒甙、毛花洋地黄甙、lopa87、vp331、rn-1-026、sg6163f、vp450和vp43。在甚至更优选的实施方案中,本发明的细胞自食增强剂选自:8-氮鸟嘌呤、米那普利二盐酸盐、lopa87、vp331和sg6163f。
它们也可以是lopa87的类似物,例如lopa90、lopa93、lopa94、lopa101、lopa104、lopa105和lopa106。
它们也可以是sg6163f的类似物,例如sg6144、sg6146和sg6149。
8-氮鸟嘌呤(c4h4n6o,嘌呤类似物)。8-氮鸟嘌呤是具有潜在抗肿瘤活性的嘌呤类似物。通过酶trna-鸟嘌呤核糖基转移酶(trna-鸟嘌呤转糖基酶)催化,8-氮鸟嘌呤与鸟嘌呤竞争而并入trna,从而干扰转运核糖核酸(trna)的修饰。改变的trna鸟嘌呤修饰涉及细胞分化和肿瘤转化。8-氮鸟嘌呤还抑制43s和80s起始复合物的形成,从而干扰翻译的起始和抑制蛋白合成。该试剂抑制肿瘤细胞生长并刺激细胞分化。
美海屈林1,5-萘二磺酸盐(c19h20n2·1/2c10h8o6s2,一种h1组胺受体拮抗剂)。美海屈林(萘二磺酸盐)是一种抗组胺药,用于由组胺释放引起的过敏症状的症状缓解,包括鼻过敏和过敏性皮肤病。
氟比洛芬(c15h13fo2,非甾体类抗炎药(nsaid)的苯基链烷酸衍生物家族)。氟比洛芬用于缓解由骨关节炎(由关节内层破裂引起的关节炎)和类风湿性关节炎(关节内层肿胀引起的关节炎)引起的疼痛、压痛、肿胀和僵硬。氟比洛芬属于一类名为nsaia(非甾体类抗炎药)的药物。它的作用是阻止身体产生导致疼痛、发烧和炎症的物质。这种丙酸类非甾族抗炎剂在结构和药理学上与非诺洛芬、布洛芬和酮洛芬有关,并且具有与其他原型nsaia类似的药理作用。氟比洛芬具有抗炎、镇痛和解热活性。市售的氟比洛芬是(+)s-和(-)r-对映体的外消旋混合物。s-对映体似乎具有大部分抗炎作用,而两种对映体都具有镇痛活性。与其他nsaia类似,氟比洛芬的抗炎作用通过环氧合酶(cox)的可逆抑制发生,环氧合酶是在前列腺素合成通路中负责将花生四烯酸转化为前列腺素g2(pgg2)以及将pgg2转化为前列腺素h2(pgh2)的酶。这有效地降低了参与炎症、疼痛、肿胀和发热的前列腺素的浓度。氟比洛芬是非选择性cox抑制剂,可抑制cox-1和-2的活性。就前列腺素抑制活性而言,它也是最有效的nsaia之一。
米那普利二盐酸盐(c17h22n4o,氨基苯基哒嗪抗抑郁药)。米那普利二盐酸盐是一种精神药物,已被证明可有效治疗各种抑郁状态。像大多数抗抑郁药一样,米那普利拮抗绝望行为。米那普利是一种氨基苯基哒嗪抗抑郁药,据报道其相对没有心脏毒性、嗜睡和体重增加。更具体地说,米那普利是一种氨基苯基哒嗪抗抑郁药,据报道其相对没有心脏毒性、嗜睡和体重增加。与其他抗抑郁治疗相似,米那普利减弱β-肾上腺素能受体功能。研究还表明,米那普利可以改善记忆巩固,重复施用药物导致这种效果的加强。此外,米那普利对记忆巩固的影响与其多巴胺能作用有关。米那普利与5-羟色胺(serotonin)2型受体和多巴胺d1和d2型受体结合。它还与5-羟色胺再摄取泵结合。因此,米那普利阻断多巴胺和5-羟色胺的再摄取。它在很小程度上也是胆碱模拟性的。因此,它可以表现出活跃情绪和促智作用。它还可作为mao-a(rima)的可逆抑制剂。还发现它可以抑制乙酰胆碱酯酶。
杨梅素(c15h10o8,苯并吡喃,小分子)。已提出磷脂酰肌醇4,5-二磷酸3-激酶催化的亚基γ同种型作为细胞靶标。
地高辛(c41h64o14,强心苷)。地高辛,一种类似于洋地黄毒甙的强心苷,用于治疗充血性心力衰竭和再入(reentry)机制导致的室上性心律失常,并在治疗慢性心房纤颤中用于控制心室率。
洋地黄毒甙(c41h64o13,强心苷)。洋地黄毒甙是一种脂溶性强心苷,可抑制质膜钠钾atp酶,导致细胞内钠和钙水平的升高、细胞内钾水平的降低。在研究中,细胞内钙的升高先于细胞死亡,并且细胞内钾的降低增加了半胱天冬酶的激活和dna片段化,引起细胞凋亡并抑制癌细胞的生长。
毛花洋地黄甙c(c49h76o20,强心苷)。毛花洋地黄甙c,从各种洋地黄中分离而得,用于治疗充血性心力衰竭和心律失常。
盐酸多柔比星(c27h29no11,癌症化疗)。盐酸多柔比星是多柔比星的盐酸盐,是一种具有抗肿瘤活性的蒽环类抗生素。多柔比星,分离自细菌波赛链霉菌青灰变种(streptomycespeucetiusvar.caesius),是柔红霉素的羟基化同源物。多柔比星嵌入dna螺旋中的碱基对之间,从而阻止dna复制并最终抑制蛋白的合成。另外,多柔比星抑制拓扑异构酶ii,其在dna复制期间产生增加的稳定的可切割酶-dna连接复合物,并且随后在双链断裂后阻止核苷酸链的连接。多柔比星还形成氧自由基,导致细胞膜脂质的脂质过氧化继发的细胞毒性;氧自由基的形成也有助于蒽环类抗生素的毒性,即心脏和皮肤血管效应。
从筛选的文库中获得的化学化合物
已从cea文库获得lopa87、vp331、rn-1026、sg6163f、vp450和vp43。还鉴定了lopa87的7种类似物(lopa90、lopa93、lopa94、lopa101、lopa104、lopa105、lopa106)和sg6163f的3种类似物(sg6144、sg6146和sg6149)。
本发明的方面
在第一方面,本发明涉及上述鉴定的化合物(在表1和表2中突出显示)用于增强癌细胞的ir-介导的细胞自食(ircce)的体外用途。
本发明的这个方面是在体外进行的。如本文所公开的,术语“体外”和“离体”是等同的并且是指使用从其通常的宿主生物(例如动物或人)中分离的生物组分(例如细胞或细胞群)进行的研究或实验。可以进一步纯化、培养或直接分析这些分离的细胞以评估突变蛋白的存在。例如,这些实验可以简化为在实验室材料中实施,例如管、烧瓶、孔、eppendorf等。相反,术语“体内”是指在整个活生物上进行的研究。
ir-介导的细胞自食可以通过任何常规手段评估。特别地,可以通过在显微镜下(通过免疫荧光或免疫组织化学)研究细胞并通过视觉检测细胞吞噬事件或细胞-内-细胞系统来评估该活性。或者,可以通过在所述细胞中测量选自以下的蛋白表达水平来评估:p53、ρ53β、ρ53γ和缺乏n-末端反式激活结构域的p53n-末端同种型(例如δ40tp53和δ133tp53);或者通过测量嘌呤能p2y2受体的表达水平或活性来评估、和/或通过测量所述细胞分泌的细胞外atp来评估,如wo2014/006227中所公开的,其通过引用并入本文。
本发明化合物也可以在体内使用。
在这种情况下,本发明的化合物优选以药物组合物的形式施用至患者,所述药物组合物还含有药学上可接受的佐剂。因此,“本发明的药物组合物”含有有效量的至少一种本发明的化合物(参见上述表1和表2)或其类似物,以及药学上可接受的佐剂或载体。非水性佐剂的示例是丙二醇、聚乙二醇、植物油和可注射的有机酯,例如油酸乙酯。其他药学上可接受的佐剂包括水溶液和无毒赋形剂,包括盐、防腐剂、缓冲剂等。静脉内载体包括液体和营养补充剂。该组合物还可包含防腐剂,例如抗微生物剂、抗氧化剂、螯合剂和惰性气体。可以根据众所周知的参数调节药物组合物各种组分的ph和精确浓度。
本发明的化合物(或本发明的组合物)的体内施用意图在将要或已经经历放射疗法治疗的受试者中增强肿瘤免疫原性。虽然不受任何特定理论的束缚,但认为这些化合物通过有助于细胞自食而暴露诱导显著保护性抗癌免疫应答的特定表位,从而与放射疗法发挥协同作用。
本公开内容旨在上述表1和表2中公开的化合物或其类似物用于制备药物组合物的用途,所述药物组合物旨在在放射疗法之前或之后施用至有此需求的受试者。
还涉及上述表1和表2中公开的化合物或其类似物用于制备药物组合物的用途,所述药物组合物意图在有此需求的受试者中加强放射疗法治疗。
最后,涉及上述表1和表2中公开的化合物或其类似物联合放射疗法用于制备用于在有此需求的受试者中治疗癌症的药物组合物中的用途。
在第二方面,本发明涉及上述表1和表2中公开的化合物或其类似物,或含有它们的任何药物组合物,用于:
·用于在患有癌症的患者中增强ir-介导的细胞自食的用途,
·用于在将接受或已接受放射疗法治疗的受试者中增强肿瘤免疫原性的用途,
·用于在将接受或已接受放射疗法治疗的受试者中诱导显著保护性抗癌免疫应答的用途,
·用于在有此需求的受试者中加强放射疗法治疗的用途,
·用于在有此需求的受试者中联合放射疗法用于治疗癌症的用途。
重要的是,施用本发明的组合物将能够减少放射的剂量,并因此导致减轻由放射治疗引起的副作用。
本发明的药物组合物可以以可注射组合物的形式(例如,静脉内、肌肉内、皮下和关节内)施用,作为液体溶液或悬浮液;也可以制备适于在注射前溶解或悬浮在液体中的固体形式。也可以乳化这些制剂。
所述药物组合物还可以通过其他途径施用,例如口服、经鼻、直肠、局部、静脉内、肌肉内、皮下、舌下、鞘内、腹膜内、关节内或皮内。优选地,施用途径是静脉内、动脉内或口服。优选地,药物组合物通过静脉内或动脉内施用。
在具体的实施方案中,当本发明的组合物用于口服施用时,本发明的组合物是片剂、溶液或胶囊,例如软凝胶胶囊。在其他具体实施方案中,当本发明的组合物用于口服施用时,其具有肠溶包衣。在其他具体实施方案中,当本发明的组合物用于口服施用时,它是油基糖浆。
在具体的实施方案中,本发明的化合物以缓释制剂施用。
本发明的组合物以足以治疗癌症的量和频率施用,并联合放射疗法治疗。受试者的进展可以通过以下来确定:测量和观察癌症标志物浓度的变化;通过测量肿瘤随时间的实际大小和/或通过确定本领域熟知的任何其他相关临床标志物。与临床进展相关的这些特征和标志物的确定、测量和评估是本领域普通技术人员所熟知的。
在一个实施方案中,本发明的组合物在放射疗法之前施用。施用时间取决于具体的制剂和细胞自食增强剂到达靶细胞所需的时间。选择施用时间以便在放射时提供细胞自食增强剂的最佳浓度。
在另一个实施方案中,在对患者施用放射疗法治疗后施用本发明的组合物。
在另一个实施方案中,本发明组合物的施用和放射疗法治疗已同时应用于患者。
本发明人观察到单独施用米那普利二盐酸盐具有对肿瘤进展的内在保护作用(参见图6b、6g、6l、6p和6q)。因此,米那普利二盐酸盐不需要与放射疗法治疗联合,它本身是有效的。这是令人惊讶的结果,因为米那普利二盐酸盐被认为是抗抑郁药,并且这种药物没有被报道过具有抗肿瘤作用。
因此,在第三方面,本发明涉及米那普利二盐酸盐,或含有它的任何药物组合物,其用于:
·用于治疗患有癌症的患者的用途,
·用于在癌症患者中诱导显著保护性抗癌免疫应答的用途。
·与放射疗法或化疗或免疫疗法治疗联合用于治疗癌症的用途。
以上公开的药物组合物的所有实施方案都可转移到该特定用途。
治疗方法
本公开还提供了在患有癌症的患者中旨在增强ir-介导的细胞自食(ircce)、在将接受或已接受放射疗法治疗的受试者中增强肿瘤免疫原性、在将接受或已接受放射疗法治疗的受试者中诱导显著保护性抗癌免疫应答、在有此需求的受试者中加强放射疗法治疗、和/或有此需求的受试者中联合放射疗法治疗癌症的方法。
在这些方法中,本发明化合物(参见表1、表2及其类似物,如上所述)或本发明的药物组合物(如上所述)在放射疗法治疗之前、之后或同时施用至所述受试者。
本发明还涉及治疗癌症患者的方法,包括向所述患者施用有效量的米那普利二盐酸盐、或含有它的任何药物组合物。事实上,所述抗抑郁药物已由本发明人显示其自身具有显著的保护性抗癌免疫应答。
筛选方法
在另一个实施方案中,本发明涉及鉴定可在受试者中加强放射疗法治疗的化合物的体外方法,所述方法包括以下步骤:
(a)将候选化合物添加到肿瘤细胞培养物中,
(b)用1-20格雷的放射剂量处理所述肿瘤细胞培养物,
(c)测量所述细胞培养物中发生的放射-介导的细胞自食,其中和对照水平相比,增强放射-介导的细胞自食的化合物能加强放射疗法治疗。
如上所述,ir-介导的细胞自食可以通过任何常规手段评估。特别地,可以通过在显微镜下(通过免疫荧光或免疫组织化学)研究细胞并通过视觉检测细胞吞噬事件或细胞-内-细胞系统来评估该活性。或者,可以通过在所述细胞中测量选自以下的蛋白表达水平来评估:p53、ρ53β、ρ53γ和缺乏n-末端反式激活结构域的p53n-末端同种型(例如δ40tp53和δ133tp53);或者通过测量嘌呤能p2y2受体的表达水平或活性来评估、和/或通过测量所述细胞分泌的细胞外atp来评估,如wo2014/006227中所公开的,其通过引用并入本文。
或者,可以在候选化合物加入细胞培养物之前,进行用放射处理细胞培养物。
在优选的实施方案中,步骤b)中使用的放射剂量在1-10格雷之间,通常为8格雷。
在优选的实施方案中,在放射发生之前或之后,肿瘤细胞培养物用候选化合物处理至少12小时,更优选至少20小时,甚至更优选24小时。
所述肿瘤细胞培养物可以是hct116细胞、ct26癌细胞、mca205纤维肉瘤细胞的培养物。
确认候选化合物的加强效果可以按照以下实施例中所述的进行,即,通过适当的肿瘤细胞培养物用化合物处理至少12小时,并应用放射处理,然后将这些细胞注入免疫活性小鼠,以确认已引发保护性免疫应答。
根据下面的实验部分,上述表1和表2中公开的化合物及其类似物在本文中称为“ir-介导的细胞自食的增强剂”的示例。
附图说明
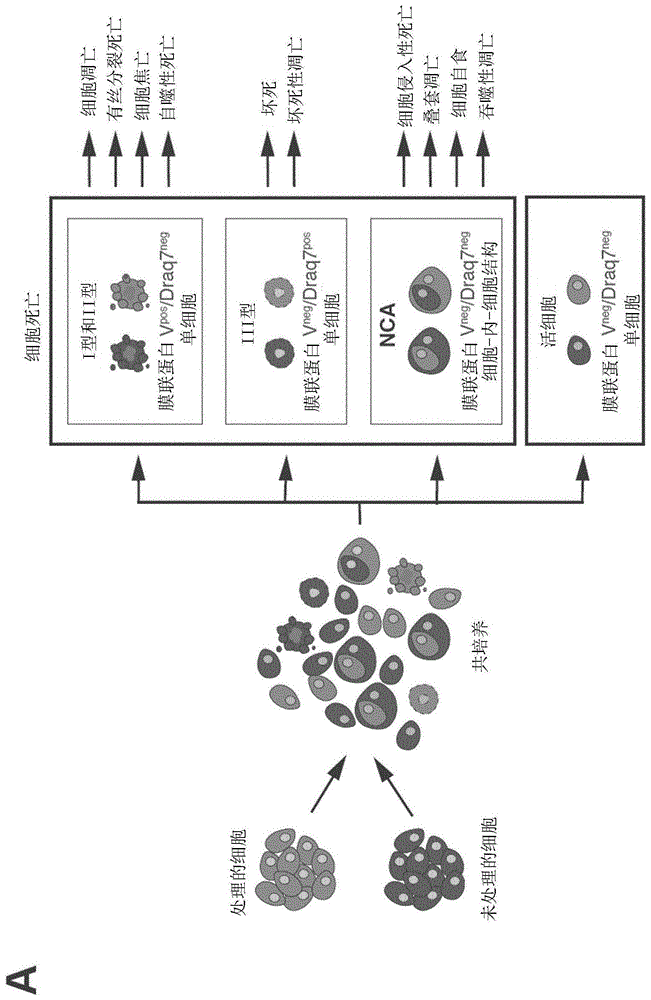
图1通过定量成像流式细胞术进行的细胞死亡谱(profiling)。(a)通过定量流式成像的细胞死亡谱的原理。(b)通过γ-辐射诱导的定量成像流式细胞术,验证细胞死亡模式的多参数和同时检测。在共培养之前,分别用cmfda(绿/浅灰)或cmtmr(红/深灰)荧光活性(vital)探针标记处理的hct116细胞和未处理的hct116细胞。共培养24小时后,分析hct116细胞的非细胞自主死亡(ncad)(通过检测cmtmr-或cmfda-标记的hct116细胞的吞噬)、磷脂酰丝氨酸(ps)暴露(使用生物素-膜联蛋白v和bv786-链霉亲和素)、血浆完整性的丧失(通过跟踪draq7摄取)和dna含量(使用hoechst33342)。使用定量流式细胞术,同时在未处理和处理的hct116细胞上检测nca死亡和典型细胞死亡(i型、ii型和iii型),将确定癌症治疗后获得的细胞死亡谱。显示了代表性图像(比例尺,20μm)。
图2通过定量成像流式细胞术进行的γ-辐射引起的细胞自主死亡模式检测。(a-e):未处理的(红/深灰)cmtmr标记的hct116细胞和未处理的(绿/浅灰)cmfda标记的hct116细胞(a、c-e)、即刻混合未处理的(红/深灰)cmtmr标记的hct116细胞和未处理的(绿/浅灰)cmfda标记的hct116细胞(c-e)共培养24小时后,或者已用4格雷γ-电离辐射照射过的未处理的(红/深灰)cmtm标记的hct116细胞和(绿/浅灰)cmfda标记的hct116细胞(绿)共培养24小时后(b-e),观察到质膜完整性丧失和ps暴露的检测和定量。在存在或不存在指定的药理学死亡效应抑制剂的情况下进行共培养。已经分析了未处理的(红/深灰)cmtmr+hct116细胞、未处理的(绿/浅灰)cmfda+hct116细胞、处理的(绿/浅灰)cmfda+hct116细胞和总细胞群(cmtmr+或cmfda+hct116细胞)的质膜完整性(用draq7)和ps暴露(用bv786-链霉亲和素/膜联蛋白v生物素)检测。显示了代表性的点图(a、b)和定量数据(c-e)(平均值±sem,n=3)。(f、g)显示未处理的(红/深灰)cmtmr+hct116细胞、未处理的(绿/浅灰)cmfda+hct116细胞、处理的(绿/浅灰)cmfda+hct116细胞、以及未处理的(红/深灰)cmtmr-标记的hct116细胞与未处理的(绿/浅灰)cmfda-标记的hct116细胞共培养24小时后获得的总细胞群(cmtmr+或cmfda+hct116细胞)(f、h-j)、即刻混合的未处理的(红/深灰)cmtmr-标记的hct116细胞与未处理的(绿/浅灰)cmfda-标记的hct116细胞(h-j)或已用4格雷γ-电离辐射照射过的未处理的(红/深灰)cmtmr标记的hct116细胞和(绿/浅灰)cmfda标记的hct116细胞(绿/浅灰)共培养24小时后(g-j)的代表性细胞周期分布。细胞周期分析的定量数据显示在(h-j)中(平均值±sem,n=3)。*或#或$代表p<0.05,##或$$代表p<0.01,***或###代表p<0.001。
图3通过定量成像流式细胞术和共聚焦荧光显微镜检测γ-辐射引起的非细胞自主死亡模式。(a-g):用4格雷γ-电离辐射照射过的未处理的(红/深灰)cmtm标记的hct116细胞和(绿/浅灰)cmfda标记的hct116细胞共培养24小时后(a)、未处理的(红/深灰)cmtmr标记的hct116细胞与未处理的(绿/浅灰)cmfda标记的hct116细胞的共培养(b、c)、或即刻混合的cmtmr标记的(红/深灰)hct116细胞和未经处理的(绿/浅灰)cmfda标记的hct116细胞(b、c),通过定量成像(a-c)和共聚焦荧光显微镜(d-g)确定细胞-内-细胞结构和靶细胞降解。如前所述,共培养后,用特异性荧光探针如bv786-链霉亲和素-膜联蛋白v生物素、draq7和hoechst33342依次标记细胞。然后,检测(红/深灰)cmtmr标记的hct116细胞内化(绿/浅灰)cmfda标记的hct116细胞(标记为r(g))和(绿色/浅灰)cmfda标记的hct116细胞内化(红/深灰)cmtmr标记的hct116细胞(标记为g(r))。代表性图像显示在(a)中(比例尺,20μm)。报道了细胞-内-细胞结构(b)和靶细胞降解(c)的频率(平均值±sem,n=3)。显示了在未处理(红)cmtmr标记的hct116细胞与未处理或γ-辐射的(绿)cmfda标记的细胞共培养期间,检测到的细胞-内-细胞结构(白色箭头)(d)和靶细胞降解(白色虚线箭头)(e)的代表性共聚焦图像(比例尺=10μm)。(f-g)确定了细胞-内-细胞结构的频率,其显示(红/深灰)cmtmr标记的细胞内化(绿/浅灰)cmfda标记的细胞(标记为r(g))、和(绿/浅灰)cmfda-标记的细胞内化(红/深灰)cmtmr标记的细胞(标记为g(r))(f),和靶细胞降解(g)(平均值±sem,n=3);#或$代表p<0.05,##或$$代表p<0.01,***或$$$或###代表p<0.001。
图4通过电离辐射触发的自食调节剂的鉴定。测试来自prestwick(a)和cea(f)文库的化合物在电离辐射后诱导细胞自食的能力。在存在或不存在10μmprestwick(a)和cea(e)文库化合物的情况下,在8格雷γ-辐射hct116细胞的同型培养物后,检测自食细胞。每个点代表一种化合物。代表性图像显示在(b,g)中。(c,h)用指定的药物处理hct116细胞。处理24小时后,通过用3,3二己氧基羰花青菁碘化物(dioc6(3))和pi染色,监测细胞死亡,并且通过流式细胞术确定垂死(dioc6(3)低ρi-,空心条)和已死(dioc6(3)低ρi+,实心条)细胞的百分比。通过荧光显微镜验证自食诱导剂。显示了代表性图像(d,i)和定量(e,j)。结果是三次测定的平均值±s.e.m.。
图5自食调节剂作为免疫原性细胞死亡诱导剂的鉴定。用指定的药物处理mca205(a)或ct26(e、i、m、q)细胞。与指定处理共培养24小时后,通过染色dioc6(3)和pi,监测细胞死亡,并且通过流式细胞术确定垂死(dioc6(3)低ρi-,空心条)和已死(dioc6(3)低ρi+,实心条)细胞的百分比。(b-d)在x辐射后,与指定化合物共培养的mca205细胞或(f-h、j-l、n-p、r-t)ct26细胞,分别皮下接种到c56bl/6或balb/c小鼠的右侧腹中。7天后,用注射到对侧腹的活细胞再次攻击小鼠,监测肿瘤生长(每组5只小鼠)。
图6肿瘤疫苗接种实验。单独用10μmvp331(a、f、k)、10μm米那普利(b、g、l)、10μmlopa87(c、h、m)、10μmsg6163f(d、i、n)、10μm氮鸟嘌呤-8(8-aza)(e、j、o)、或与8gy电离辐射联合对ct26细胞处理24小时,并接种(s.c.)到免疫活性的balb/c小鼠中,7天后采用相同的癌细胞在对侧腹再次攻击。在接下来的38天,每周三次评价无肿瘤小鼠的百分比。显示了完全无肿瘤小鼠(a-e)、第一注射位点(f至j,p)或第二注射位点(k至o,q)的无肿瘤小鼠的百分比(*p<0.05,**p<0.01,***p<0.001,双因素anova)。
具体实施方式
实施例
1.材料和方法
化学品、细胞系和培养条件
除非另有说明,否则化学品购自sigma-aldrich。抗生素、介质、细胞培养补充剂获自lifetechnologies。苄氧基羰基-val-ala-asp(ome)-氟甲基酮(z-vad-fmk)来自bachem和重组小鼠tnf-α来自r&d系统。人结肠癌hct116细胞在mccoy's5a介质中培养,鼠纤维肉瘤细胞系l929在dulbecco改良eagle介质中培养。所有介质补充有10%热灭活的胎牛血清(fbs)、10mmhepes缓冲液、2mml-谷氨酰胺、10u/ml青霉素钠和10μg/ml硫酸链霉素。
放射
将细胞接种在6孔板、12孔板或25cm2的烧瓶中,并用指定剂量的γ射线放射器ibl-637(cs137,1gy/min,γcis-biointernational,iba,saclay,法国)进行放射。
celltrackertm荧光探针标记
除去培养介质后,将hct116细胞与预热的介质在37℃孵育45分钟,所述介质含有10μm的5-氯甲基荧光素二乙酸酯(cmfda,绿色荧光)或5-(和-6)-(((4-氯甲基)苯甲酰基)氨基)四甲基罗丹明(cmtmr,红色荧光)(molecularprobes-lifetechnologie)。此后,用预热的介质冲洗hct116细胞两次,并在37℃孵育1小时。如所示,处理染色的细胞,并培养以进行细胞死亡谱分析。
通过定量流式成像进行细胞死亡谱
用cmfda(绿色荧光,cmfda+)或cmtmr(红色荧光,cmtmr+)标记未处理的hct116细胞,并用cmfda(绿色荧光,cmfda+)标记处理的hct116细胞。进行以下细胞混合:将未处理的cmtmr+hct116细胞与未处理的cmfda+hct116细胞混合,或将未处理的cmtmr+hct116细胞与处理的cmfda+hct116细胞混合。然后,细胞在存在或不存在rock的药理学抑制剂(y27632(30μm))、泛半胱天冬酶抑制剂(z-vad-fmk(zvad,100μm))、半胱天冬酶-1抑制剂(ac-yvad-cmk(yvad,100μm))、坏死抑制剂(necrostatin-1(nec1、30μm))、抑制自噬的液泡型h(+)-atp酶抑制剂(v-atp酶)(巴弗洛霉素al(bafa1,50nm))、具有抗有丝分裂活性的cdks抑制剂(roscovitine(rosco,10μm))的情况下,共培养24小时。共培养24小时后,收集分离的和贴壁的细胞,并在37℃在温热的完全介质中用hoechst33345(10μg/ml)染色1小时。为了检测磷脂酰丝氨酸(ps)暴露和质膜通透性,标记的hct116细胞依次与制造商推荐的生物素-膜联蛋白v(bdpharmingen)、0.5μg的bv786-链霉亲和素(bdbiosciences)和3μm的draq7(biostatus)在25℃一起孵育15分钟。用pbs溶液洗涤后,立即使用成像流式细胞仪
流式细胞术和共聚焦荧光显微镜
为了检测ps暴露、质膜通透性和细胞周期进程,在共培养后,细胞依次用特异性荧光探针(例如fitc-缀合的膜联蛋白v、碘化丙啶和hoechst33342)标记,并通过流式细胞术分析。收集分离的和贴壁的细胞,并在37℃在温热的完全介质中用hoechst33345(10ug/ml)染色1小时。用pbs洗涤后,按照制造商的说明,将hct116细胞悬浮于补充有异硫氰酸酯荧光素(fitc)-缀合的膜联蛋白v(bdbiosciences)和碘化丙啶(pi,1μg/ml)(sigma)的1×结合缓冲液中。然后使用lsrii流式细胞仪(bectondickinson)和flowjo软件v10分析样品。对于共聚焦荧光显微术,在3.7%多聚甲醛-pbs中共培养15分钟后,固定hct116细胞,并用1μg/mlhoechst33342(invitrogen)复染15分钟。然后,通过配备有apochromat63×1.3na和63×1.15na油浸物镜的共聚焦spe显微镜分析细胞。使用leicaaplicationsuite(las)软件(leicamicrosystems)。
western印迹
在裂解缓冲液(含有1%np40、20mmol/lhepes、10mmol/lkcl、1mmol/ledta、10%甘油、蛋白酶和磷酸酶抑制剂片剂)中提取总细胞蛋白。将蛋白提取物(30μg)在4-12%
统计分析
每个实验至少重复三次,产生可比较的结果。除非另有说明,否则附图示出了来自一个代表性实验的定量数据(平均值±sem,n=3)。通过prismv.5.03(graphpadsoftware,lajolla,ca,usa)分析数据。通过双尾学生t检验评估统计显著性。在所有实验中,p值<0.05被认为是统计显著的。
ii.结果
使用多光谱成像流式细胞术进行的细胞死亡谱分析允许同时检测非细胞自主和细胞自主死亡模式。细胞通过nca过程死亡的能力使我们从概念的角度重新考虑我们考虑细胞死亡过程的方法。实际上,要考虑的形态学和/或生化参数选择以及用于检测细胞死亡的技术方法事先预确定了预期的结果,并且不允许或很少鉴定新的致死过程,例如细胞自食或细胞侵入性死亡。放射疗法领域也面临着这个问题。实际上,已经在不同的研究中揭示了放射可以引发许多致命过程,例如细胞凋亡、自噬性细胞死亡、坏死或有丝分裂死亡35,36。最近在另外的研究中已经表明,用相同剂量对相同细胞类型的放射可以引发细胞凋亡37,但也可以引发有丝分裂死亡38。在先前的研究中,发现在放射后非常快速地观察到细胞凋亡和有丝分裂死亡的发生,这与在较长期内观察到的克隆形成存活率无关35。这些研究强调了消除放射细胞所涉及的未知致死过程的存在。此外,越来越多的出版物揭示了细胞自食和细胞侵入性死亡在控制肿瘤生长和消除转移细胞方面的影响,促使人们追踪ncad这一过程的开始。
考虑到可能引发非细胞自主和细胞自主细胞死亡模式的致死刺激的多样性、以及控制两种过程的信号传导通路的复杂性(涉及(或不涉及)半胱天冬酶、组织蛋白酶或颗粒酶),决定同时检测ir引发的非细胞自主和细胞自主死亡模式。为了确定致死性损伤后细胞群是否可以同时经历按照细胞自主或非细胞自主方式执行的直接或/和旁路的细胞杀伤,基于hct116细胞和同基因hct116细胞的共培养设计了细胞死亡谱分析,所述hct116细胞已经用5-氯甲基荧光素二乙酸酯(cmfda,绿)荧光活性探针进行标记并通过电离辐射(γ射线)进行处理,所述同基因hct116细胞已经用5-(和-6)-(((4-氯甲基)苯甲酰基)氨基)四甲基罗丹明(cmtmr,红)荧光活性探针进行标记。共培养24小时后,分析经处理的cmfda+细胞、未处理的cmtmr+细胞和总细胞群(cmfda+细胞和cmtmr+细胞)的磷脂酰丝氨酸(ps)暴露、质膜完整性丧失和dna含量,以便同时检测处理的细胞和(未处理的)邻近细胞的细胞死亡诱导。为了表征在执行检测到的细胞死亡模式中所涉及的分子机制,在存在细胞死亡调节剂(比如,rock1抑制剂(y27632)、泛半胱天冬酶抑制剂(zvad-fmk)、半胱天冬酶-1抑制剂(yvad-fmk)、坏死抑制剂(nec1)、巴弗洛霉素al(bafa1)和roscovitine(rosco))的情况下进行共培养,上述调节剂已知分别可抑制细胞吞噬(启动细胞侵入性死亡16、叠套凋亡17和细胞自食16的过程)、半胱天冬酶-3或半胱天冬酶-7(有助于执行细胞凋亡39、有丝分裂死亡38,40或叠套凋亡17)或半胱天冬酶-1(细胞焦亡所需的41)的蛋白水解切割、促坏死性凋亡的激酶rip1激酶(ripk1)的激活(有助于坏死性凋亡42)、自噬体和溶酶体之间的融合(因此损害自噬和自噬相关细胞死亡过程中的自噬泡成熟43)、以及最后,细胞周期蛋白依赖性激酶1(cdk1)-细胞周期蛋白b的活性和通过有丝分裂的进展(这是有丝分裂相关死亡所需的,比如有丝分裂死亡40,44)。同时检测ps暴露、质膜完整性的丧失、细胞伴侣的dna含量以及细胞死亡调节剂的药理学抑制,使我们能够在共培养期间在靶细胞和邻近细胞上,通过使用多光谱成像流式细胞仪检测至少9种进行的细胞死亡模式(包括细胞凋亡、有丝分裂死亡、细胞焦亡、自噬细胞死亡、坏死、坏死性凋亡、细胞侵入性死亡、叠套凋亡、和细胞自食)(图1,a-b),从而区分非细胞自主死亡和细胞自主死亡,以及区分直接细胞杀死和旁路致死效应。该方法允许人们定义由ir引发的细胞死亡谱。
电离辐射引发的细胞死亡谱突出了在经放射的和非放射的癌细胞上诱导细胞死亡。尽管实施了密集的生物和药物研究以更好地表征与抗癌治疗相关的细胞和生物化学过程,但是作为临床中最常用的抗癌治疗之一的放射疗法,其治疗效果的致死机制仍然是未知的。已经在响应于电离辐射中检测到的致死过程(例如细胞凋亡和有丝分裂障碍)并未直接涉及治疗效果35,这表明仍然未知的其他细胞死亡模式可能有助于放射疗法的治疗效果。在此背景下确定了经放射的癌细胞的细胞死亡谱。根据上述方法,cmfda-标记的细胞用4格雷进行放射或不放射,24小时后以1:1的比例与cmtmr标记的细胞混合,并在每种指定的抑制剂存在下培养24小时(补充图1a-1e)。然后,使用膜联蛋白v-bv786、draq7和hoechst33342染色分别确定ps暴露、每种细胞伴侣的膜完整性和dna含量。尽管在邻近细胞群(cmtmr+细胞)(图2a、2b和2d)和未处理的对照细胞群(图2a、2d和2e)上没有观察到细胞凋亡和坏死性细胞死亡的显著增加(如膜联蛋白v+draq7-和draq7+细胞的分析所揭示的),在共培养24小时后,在总细胞群上(通过考虑cmfda+和cmtmr+细胞群显示的)(图2a-2c)和放射的细胞群(cmfda+细胞)(图2b和2e)上检测到这两种类型的死亡显著增加,证明本方法允许检测非放射和放射的细胞的细胞死亡模式。还观察到泛半胱天冬酶抑制剂z-vad-fmk和药理学细胞周期蛋白依赖性激酶抑制剂roscovitine(rosco)抑制在放射的cmfda+细胞外质膜上的ps暴露(图2e),证实了先前发表的45,46经放射的cmfda+细胞需要半胱天冬酶的激活和经有丝分裂而发展至死亡。此外,巴弗洛霉素a1(bafa1)对自噬通量的损害增加了总细胞群(cmfda+和cmtmr+细胞)(图2c)、非-放射的cmtmr+细胞(图2d)和放射的cmfda+细胞(图2e)中垂死细胞(膜联蛋白v+draq7-和draq7+细胞)的频率,从而揭示自噬是通过电离辐射引发的存活细胞机制,其有助于拯救非放射的和放射的细胞免于死亡。此外,通过细胞周期同时分析处理的和未处理的细胞的进展表明,细胞死亡诱导与放射的cmfda+细胞逃离g1停滞相关,并导致它们在s期和g2/m期累积(图2f、2g和2j)。在总体或cmtmr+细胞群上未检测到细胞周期的改变,表明仅在放射的细胞上检测到细胞周期改变(图2f、2g和21)。经典流式细胞仪分析也证实了这些结果(补充图2a-2b),证明在电离辐射后,放射的和非放射的癌细胞都经历半胱天冬酶-1依赖性细胞死亡。总之,这些结果突出了抗癌剂通过直接细胞杀伤和旁路效应同时消除癌细胞的能力。
电离辐射引发的细胞死亡谱也揭示了非细胞自主死亡模式的诱导。同时,在相同的共培养物中确定了放射的cmfda+细胞吞噬或侵入邻近细胞的能力,诱导细胞自食相关的细胞死亡所需的两个细胞过程(如细胞自食、叠套凋亡或吞噬性凋亡)或细胞-内-细胞侵袭引发的细胞死亡(如细胞侵入性死亡)。多光谱成像流式细胞术分析显示γ-辐射的cmfda+细胞触发了邻近细胞的吞噬(如通过γ-辐射的cmfda+细胞对“靶”cmtmr+细胞的内化所揭示的(图3a和3b))。不受泛半胱天冬酶抑制剂z-vad-fmk影响的这一过程受到rock1抑制剂(y27632)的抑制(图3b),从而揭示了检测到的细胞-内-细胞内化不同于凋亡细胞的吞噬摄取,并且需要rock1活性发生。通过共聚焦显微镜也证实了γ-辐射的细胞的自食活性(图3d-3f)。有趣的是,细胞死亡谱分析还允许区分活细胞吞噬和活cmtmr+细胞对凋亡cmfda+细胞的吞噬作用,这与用巴弗洛霉素a1处理而诱导细胞凋亡是连续的(图3b)。然后,评估吞噬的cmtmr+细胞和放射的吞噬cmfda+细胞的细胞命运。观察到几乎所有吞噬的cmtmr+细胞都显示出细胞降解的迹象(如通过多光谱成像流式细胞术(图3a)和共聚焦显微镜(图3d和3e)检测到的内化细胞dna含量损失所揭示的)。在泛半胱天冬酶抑制剂z-vad-fmk和半胱天冬酶-1抑制剂yvad-fmk存在的情况下,该过程显著减少,揭示了ir介导的吞噬细胞死亡的发生需要半胱天冬酶并且可以通过半胱天冬酶-1依赖性细胞凋亡(也称为细胞焦亡)来进行。此外,还发现90%的自食细胞未暴露ps或表现出质膜完整性的丧失(补充图2c),强调在ir介导细胞吞噬后,内化细胞沉淀而死亡,没有调节自食细胞的活力。总之,这些结果表明,通过直接细胞杀伤、旁路致死效应和细胞自食相关的细胞死亡的综合作用,电离辐射同时消除癌细胞。这些结果突出了迫切需要同时测量致死过程中非细胞自主和细胞自主死亡子程序的诱导。
化学文库筛选导致ir-介导的细胞自食增强剂的鉴定。然后,开发了化学化合物文库的筛选,以鉴定能够诱导ir介导的细胞自食的化合物。因此,用8格雷放射剂量处理的hct116细胞,用橙色cmtmr细胞示踪剂或绿色cmfda细胞示踪剂染色,并在10μm化学化合物存在下培养。培养24小时后,细胞用核染料(5μg/mlhoechst33342在37℃下1小时)染色,并使用
ircce与ir的组合诱导有效的抗肿瘤免疫。
考虑到免疫原性细胞死亡(icd)诱导剂(如地高辛、洋地黄毒甙、毛花洋地黄甙c或盐酸多柔比星3,4,48-51)被鉴定为ircce,评估放射疗法后这些化合物诱导抗肿瘤免疫力的能力。首先,开发了临床前方法来研究ircce对两种小鼠癌模型(结肠ct26癌和纤维肉瘤mca205)的免疫学作用。最初,通过抗肿瘤疫苗接种测定法使用免疫活性小鼠来理解ircce+ir组合触发特异性抗肿瘤免疫应答的能力。根据先前发表的研究52,将屈从于免疫原性细胞死亡(icd)的癌细胞注射到免疫活性小鼠中必然引发对肿瘤抗原特异性的保护性免疫应答。因此,首先3×105mca205细胞用10μm的sg6163f处理24小时。然后,将处理过的细胞在200μlpbs中皮下接种到8周龄雌性c57bl/6小鼠的下腹侧。一周后,将3×104个未处理的对照细胞接种到小鼠的对侧腹侧。使用普通卡尺每周评估肿瘤。携带超过20-25%体重的肿瘤的动物被安乐死。观察到单独用ir或sg6163f处理的小鼠不能在体外诱导细胞死亡的显著增加(图5a)。事实上,它们在接种未处理的细胞后没有表现出保护性应答,这表明放射剂量和所用sg6163f的浓度不足以刺激抗癌免疫应答(图5b)。这些结果与先前公布的结果一致,证明仅在癌细胞暴露于体外细胞毒性抗癌处理时才检测到保护性应答,直到70%的细胞在质膜的外部小叶上暴露磷脂酰丝氨酸48。令人惊讶的是,观察到用8gyir和10μmsg6163f组合处理mca205细胞在已注射的5只小鼠的3只中诱导了保护性应答(图5b)。这些结果首次揭示了ircce+ir的组合可以在没有显著增加处理和放射的细胞死亡的情况下引发ir介导的抗癌免疫应答(图5a),表明细胞自食或细胞自食相关的信号传导通路可能有助于诱导肿瘤免疫原性。然后,在10μm的sg6163f(图5e和5f)、vp331(图51和5j)、米那普利二盐酸盐(图5m和5n)或lopa87(图5q和5r)存在下,3×106ct26细胞以8gy放射24小时。然后,如先前所述,在200μlpbs中将细胞皮下接种到8周龄雌性balb/c小鼠的下腹侧。一周后,将5×105个未处理的对照细胞接种到小鼠的对侧腹侧中,并使用普通卡尺每周评估肿瘤。如前所述,携带超过20-25%体重肿瘤的动物被安乐死。通过用dioc(6)3/ip染色(图5e、5i、5m和5q)测定,来评估每种条件下垂死细胞的百分比。最后,明确了这些化合物抑制癌细胞生长的能力,所述癌细胞生长是注射放射的和治疗的癌细胞后7天注射的。观察到,在注射已经用ir和化学化合物处理的癌细胞后显示保护性应答的小鼠频率显著增加(图5h、5l、5p和5t)。
如前所述,在ct26小鼠癌模型上进行第二抗肿瘤疫苗接种测定。简而言之,在10μm的vp331(图6a、6f、6k)、米那普利二盐酸盐(图6b、6g、6l)、lopa87(图6c、6h、6m)、sg6163f(图6d、6i、6n)、或氮鸟嘌呤-8(8-aza)(图6e、6j、6o)存在时,ct26细胞用8gy放射24小时。然后,如前所述将细胞接种到免疫活性balb/c小鼠中。最后,明确了这些化合物抑制癌细胞生长的能力,所述癌细胞是注射放射的和处理的癌细胞后7天注射的。证实了之前的结果。观察到,在注射已经用ir和化学化合物处理的癌细胞后显示保护性应答的小鼠频率显著增加(图6p和6q)。有趣的是,单独的米那普利二盐酸盐(图6b、6g、6l、6p和6q)和氮鸟嘌呤-8(图6e、6j、6o、6p和6q)诱导注射癌细胞后保护性应答的增加。
总而言之,这些结果揭示了用本发明平台鉴定的化学化合物诱导保护性抗癌免疫应答的能力。
参考文献
1tobiasjs.clinicalpracticeofradiotherapy.lancet1992;339∶159-163.
2apetohl,ghiringhellif,tesniereaetal.theinteractionbetweenhmgb1andtlr4dictatestheoutcomeofanticancerchemotherapyandradiotherapy.immunolrev2007;220∶47-59.
3apetohl,ghiringhellif,tesnicreaetal.toll-likereceptor4-dependentcontributionoftheimmunesystemtoanticancerchemotherapyandradiotherapy.naturemedicine2007;13∶1050-1059.
4ghiringhellif,apetohl,tesniereaetal.activationofthenlrp3inflammasomeindendriticcellsinducesil-1beta-dependentadaptiveimmunityagainsttumors.naturemedicine2009;15∶1170-1178.
5michaudm,martinsi,sukkurwalaaqetal.autophagy-dependentanticancerimmuneresponsesinducedbychemotherapeuticagentsinmice.science2011;334∶1573-1577.
6kingsleydp.aninterestingcaseofpossibleabscopaleffectinmalignantmelanoma.thebritishjournalofradiology1975;48∶863-866.
7ohbak,omagarik,nakamuratetal.abscopalregressionofhepatocellularcarcinomaafterradiotherapyforbonemetastasis.gut1998;43∶575-577.
8postowma,callahanmk,barkercaetal.immunologiccorrelatesoftheabscopaleffectinapatientwithmelanoma.thenewenglandjournalofmedicine2012;366∶925-931.
9reesgj,rosscm.abscopalregressionfollowingradiotherapyforadenocarcinoma.thebritishiournalofradiologv1983∶56∶63-66.
10demarias,ngb,devittmletal.ionizingradiationinhibitionofdistantuntreatedtumors(abscopaleffect)isimmunemediated,internationaljournalofradiationoncology,biology,physics2004;58∶862-870.
11mengy,beckettma,lianghetal.blockadeoftumornecrosisfactoralphasignalingintumor-associatedmacrophagesasaradiosensitizingstrategy.cancerresearch2010;70∶1534-1543.
12dewanmz,gallowayae,kawashimanetal.fractionatedbutnotsingle-doseradiotherapyinducesanimmune-mediatedabscopaleffectwhencombinedwithanti-ctla-4antibody.clinicalcancerresearch:anofficialjournaloftheamericanassociationforcancerresearch2009;15∶5379-5388.
13kroemerg,galluzzil,vandenabeelepetal.classificationofcelldeath:recommendationsofthenomenclaturecommitteeoncelldeath2009.celldeathanddifferentiation2009;16∶3-11.
14vandenberghet,vanlangenakkcrn,parthoenseetal.necroptosis,necrosisandsecondarynecrosisconvergeonsimilarcellulardisintegrationfeatures.celldeathanddifferentiation2010;17∶922-930.
15aaestl,kaczmareka,delvaeyetetal.vaccinationwithnecroptoticcancercellsinducesefficientanti-tumorimmunity.cellreports2016;15∶274-287.
16overholtzerm,mailleuxaa,mouneimnegetal.anonapoptoticcelldeathprocess,entosis,thatoccursbycell-in-cellinvasion.cell2007;131∶966-979.
17wangs,hemf,chenyhetal.rapidreuptakeofgranzymebleadstoemperitosis:anapoptoticcell-in-celldeathofimmunekillercellsinsidetumorcells.celldeath&disease2013;4:e856.
18browngc,neherjj.eatenalive!celldeathbyprimaryphagocytosis:′phagoptosis′.trendsinbiochemicalsciences2012;37∶325-332.
19browngc,vilaltaa,frickerm.phagoptosis-celldeathbyphagocytosis-playscentralrolesinphysiology,hostdefenseandpathology.currentmolecularmedicine2015;15∶842-851.
20sierrof,tayss,warrenaetal.suicidalemperipolesis:aprocessleadingtocell-in-cellstructures,tcellclearanceandimmunehomeostasis.currentmolecularmedicine2015;15∶819-827.
21khandelwals,vanrooijenn,saxenark.reducedexpressionofcd47duringmurineredbloodcell(rbc)senescenceanditsroleinrbcclearancefromthecirculation.transfusion2007;47∶1725-1732.
22lagassee,weissmanil.bcl-2inhibitsapoptosisofneutrophilsbutnottheirengulfmentbymacrophages.thejournalofexperimentalmedicine1994;179∶1047-1052.
23overholtzerm,bruggejs.thecellbiologyofcell-in-cellstructures.natrevmolcellbiol2008;9∶796-809.
24liy,sunx,deysk.entosisallowstimelyeliminationoftheluminalepithelialbarrierforembryoimplantation.cellreports2015;11∶358-365.
25benselerv,warrena,vometal.hepatocyteentryleadstodegradationofautoreactivecd8tcells.procnatlacadsciusa2011;108∶16735-16740.
26nic,huangl,chenyetal.implicationofcell-in-cellstructuresinthetransmissionofhivtoepithelialcells.cellresearch2015;25∶1265-1268.
27nic,cheny,zengmetal.in-cellinfection:anovelpathwayforepstein-bartvirusinfectionmediatedbycell-in-cellstructures.cellresearch2015;25∶785-800.
28bartoshtj,ullahm,zeitounis,beaverj,prockopdj.cancercellsenterdormancyaftercannibalizingmesenchymalstem/stromalcells(mscs).procnatlacadsciusa2016;113:e6447-e6456.
29luginil,matarresep,tinariaetal.cannibalismoflivelymphocytesbyhumanmetastaticbutnotprimarymelanomacells.cancerresearch2006;66∶3629-3638.
30wangs,guoz,xiapetal.internalizationofnkcellsintotumorcellsrequiresezrinandleadstoprogrammedcell-in-celldeath.cellresearch2009;19∶1350-1362.
31hemf,wangs,wangy,wangxn.modelingcell-in-cellstructureintoitsbiologicalsignificance.celldeath&disease2013;4∶e630.
32sunq,cibases,huangh,hodgsonl,overholtzerm.inductionofentosisbyepithelialcadherinexpression.cellresearch2014;24∶1288-1298.
33canoce,sandimj,hamiditetal.homotypiccellcannibalism,acell-deathprocessregulatedbythenuclearprotein1,opposestometastasisinpancreaticcancer.embomolecularmedicine2012;4∶964-979.
34sunq,luot,renyetal.competitionbetweenhumancellsbyentosis.cellresearch2014;24∶1299-1310.
35abendm.reasonstoreconsiderthesignificanceofapoptosisforcancertherapy.internationaljournalofradiationbiology2003;79∶927-941.
36gudkovav,komarovaea.theroleofp53indeterminingsensitivitytoradiotherapy.naturereviewscancer2003;3∶117-129.
37zhangp,castedom,taoyetal.caspaseindependenceofradio-inducedcelldeath.oncogene2006;25∶7758-7770.
38castedom,perfettinijl,roumiert,andreauk,medemar,kroemerg.celldeathbymitoticcatastrophe:amoleculardefinition.oncogene2004;23∶2825-2837.
39garcia-calvom,petersonep,leitingb,ruelr,nicholsondw,thornberryna.inhibitionofhumancaspasesbypeptide-basedandmacromolecularinhibitors.thejournalofbiologicalchemistry1998;273∶32608-32613.
40castedom,perfettinijl,roumiertetal.thecellcyclecheckpointkinascchk2isanegativeregulatorofmitoticcatastrophe.oncogene2004;23∶4353-4361.
41miaoea,leafia,treutingpmetal.caspase-1-inducedpyroptosisisaninnateimmuneeffectormechanismagainstintracellularbacteria.natureimmunology2010;11∶1136-1142.
42degtereva,huangz,boycemetal.chemicalinhibitorofnonapoptoticcelldeathwiththerapeuticpotentialforischemicbraininjury.naturechemicalbiology2005;1∶112-119.
43yamamotoa,tagaway,yoshimorit,moriyamay,masakir,tashiroy.bafilomycina1preventsmaturationofautophagicvacuolesbyinhibitingfusionbetweenautophagosomesandlysosomesinrathepatomacellline,h-4-ii-ecells.cellstructureandfunction1998;23∶33-42.44meijerl,borgnea,mulneroetal.biochemicalandcellulareffectsofroscovitine,apotentandselectiveinhibitorofthecyclin-dependcntkinasescdc2,cdk2andcdk5.europeanjournalofbiochemistry1997;243∶527-536.
45lowesw,schmittem,smithsw,osborneba,jackst.p53isrequiredforradiation-inducedapoptosisinmousethymocytes.nature1993;362∶847-849.
46vakifahmetogluh,olssonm,zhivotovskyb.deaththroughatragedy:mitoticcatastrophe.celldeathanddifferentiation2008;15∶1153-1162.
47zhangxd,yangxc,chungnetal.robuststatisticalmethodsforhitselectioninrnainterferencehigh-throughputscreeningexperiments.pharmacogenomics2006;7∶299-309.
48mengerl,vacchellie,adjemiansetal.cardiacglycosidesexertanticancereffectsbyinducingimmunogeniccelldeath.sciencetranslationalmedicine2012;4∶143ra199.
49obeidm,panaretakist,jozanetal.calreticulinexposureisrequiredfortheimmunogenicityofgamma-irradiationanduvclight-inducedapoptosis.celldeathanddifferentiation2007;14∶1848-1850.
50obeidm,tesnierea,ghiringhellifetal.calreticulinexposuredictatestheimmunogenicityofcancercelldeath.naturemedicine2007;13∶54-61.
51casaresn,pequignotmo,tesniereaetal.caspase-dependentimmunogenicityofdoxorubicin-inducedtumorcelldeath.thejournalofexperimentalmedicine2005;202∶1691-1701.
52greendr,fergusont,zitvogell,kroemerg.immunogenicandtolerogeniccelldeath.natrevimmunol2009;9∶353-363.
- 还没有人留言评论。精彩留言会获得点赞!