蓝芩口服液指纹图谱的建立方法及其指纹图谱和应用与流程
本发明涉及药物分析领域,具体涉及一种蓝芩口服液指纹图谱的建立方法及其指纹图谱和应用。
背景技术:
蓝芩口服液是扬子江药业集团根据临床验方自主开发而成的纯中药制剂,由板蓝根、黄芩、黄柏、栀子、胖大海五味中药组成,经药理药效研究和临床试验证明具有显著的解热、镇痛、抗炎、抗菌、抗病毒五效合一的作用,主要用于急性咽炎,肺胃实热证所致的咽痛、咽干、咽部灼热等症,对上呼吸道感染等有明显的抗病毒、抗菌消炎作用,尤其在退热、消除咽痛、咳嗽等症状方面,疗效确切。蓝芩口服液因其疗效广,不良反应少,而且使用人群较广,越来越受到社会的重视,发展前景十分广大。
药品的质量是保证临床用药安全有效的前提,目前常用来评价蓝芩口服液制剂质量的方法是对栀子苷含量进行控制,但这种方法不能从整体上反映中医用药的整体疗效。要控制中成药的质量,需要对物质群整体予以控制。
技术实现要素:
以下是对本文详细描述的主题的概述。本概述并非是为了限制权利要求的保护范围。
本发明的目的是提供一种蓝芩口服液指纹图谱的建立方法及其指纹图谱和应用,通过该方法建立蓝芩口服液及其关键工艺中间体包括浓缩工艺中间体i、收膏工艺中间体ii、初配工艺中间体iii、精配工艺中间体iv的指纹图谱,并可以对蓝芩口服液及其关键工艺中间体中多个药材的多个药效成分进行定性定量分析,根据不同工艺过程中含量的变化规律,对制剂不同工艺环节的质量控制,对于全过程质量控制体系的建立提供了一定的理论依据和支持,能较全面的反映蓝芩口服液的质量信息,保证产品质量均一稳定。
在本发明的实施方案中,本发明提供了一种蓝芩口服液指纹图谱的建立方法,所述建立方法包括以下步骤:
(1)供试品溶液的制备:以蓝芩口服液制备得供试品溶液;
(2)对照品溶液的制备:以溶解有腺苷对照品、表告依春对照品、盐酸黄柏碱对照品、绿原酸对照品、木兰花碱对照品、栀子苷对照品、苯甲酸对照品、盐酸小檗碱对照品、黄芩苷对照品、汉黄芩苷对照品、黄芩素对照品和汉黄芩素对照品的溶液为对照品溶液;
(3)采用hplc分别测定步骤(1)的供试品溶液和步骤(2)的对照品溶液,并分别得到所述供试品溶液和所述对照品溶液的液相色谱;
(4)将步骤(3)得到的液相色谱导入中药色谱指纹图谱相似度评价系统分析,得到蓝芩口服液的指纹图谱。
在本发明的实施方案中,步骤(1)中所述蓝芩口服液可以用蓝芩口服液关键工艺中间体代替;所述蓝芩口服液关键工艺中间体包括浓缩工艺中间体i、收膏工艺中间体ii、初配工艺中间体iii、精配工艺中间体iv;而且,所述蓝芩口服液及其关键工艺中间体样品来自生产的不同批次的产品。
蓝芩口服液的制备工艺为:取板蓝根300.0g,黄芩240.0g,栀子300.0g,黄柏120.0g,胖大海100.0g,以上五味,加水煎煮三次,第一次2小时,第二次、第三次各1小时。合并煎煮液,滤过,滤液浓缩至相对密度为1.10-1.12(70℃)。加乙醇使含醇量达60%左右,静置12小时,滤过。10℃以下静置24小时,滤过,加灌装,灭菌,即得。其中“滤液浓缩至相对密度为1.10-1.12(70℃)”为浓缩工艺中间体;“滤液减压回收乙醇并浓缩至相对密度为1.15-1.20(60℃)”为收膏工艺中间体ii、“另取蔗糖300g,制成糖浆,与上述药液合并,加热煮沸15分钟,加苯甲酸钠3g,”为初配工艺中间体iii;“0.3%的聚山梨酯80,加水至1000ml,混匀”为精配工艺中间体iv。
在本发明的蓝芩口服液指纹图谱的建立方法的实施方案中,其中,所述步骤(1)的供试品溶液的制备包括:精密吸取蓝芩口服液100~300μl于10ml离心管中,加入4~8ml的体积分数为50%-100%的甲醇水溶液进行稀释,混匀后过0.45μm微孔滤膜。作为优选方案,所述步骤(1)供试品溶液的制备包括:精密吸取蓝芩口服液200μl于10ml离心管中,加入4.8ml的甲醇进行稀释,混匀后过0.45μm微孔滤膜。
在本发明的蓝芩口服液指纹图谱的建立方法的实施方案中,其中,所述步骤(2)的对照品溶液的制备包括:精密称取腺苷对照品、表告依春对照品、盐酸黄柏碱对照品、绿原酸对照品、木兰花碱对照品、栀子苷对照品、苯甲酸对照品、盐酸小檗碱对照品、黄芩苷对照品、汉黄芩苷对照品、黄芩素对照品和汉黄芩素对照品,加入体积分数为70%-100%的甲醇水溶液制成每1ml含有5~8μg腺苷、2~6μg表告依春、5~8μg盐酸黄柏碱、30~50μg绿原酸、2~5μg木兰花碱、200~300μg栀子苷、100~200μg苯甲酸、2~5μg盐酸小檗碱、150~250μg黄芩苷、50~100μg汉黄芩苷、8~15μg黄芩素、8~15μg汉黄芩素的对照品溶液。作为优选方案,所述步骤(2)对照品溶液的制备包括:取黄芩苷对照品适量,精密称定,加体积分数为70%甲醇水溶液溶解,配制成含量为208μg·ml-1的对照品溶液,分别取腺苷对照品、表告依春对照品、盐酸黄柏碱对照品、绿原酸对照品、木兰花碱对照品、栀子苷对照品、苯甲酸对照品、盐酸小檗碱对照品、汉黄芩苷对照品、黄芩素对照品和汉黄芩素对照品适量,精密称定,加甲醇溶解,配制成含量分别为7.52μg·ml-1、3.84μg·ml-1、6.44μg·ml-1、32.96μg·ml-1、2.52μg·ml-1、284.00μg·ml-1、162.00μg·ml-1、4.56μg·ml-1、98.00μg·ml-1、10.60μg·ml-1和9.76μg·ml-1的对照品溶液。
在本发明的蓝芩口服液指纹图谱的建立方法的实施方案中,其中,所述步骤(3)包括设置hplc色谱条件:色谱柱采用十八烷基硅烷键合硅胶为填料,柱温25-35℃,流速为0.8-1.2ml/min,检测波长250-320nm,并开启dad检测;以乙腈为流动相a,以体积分数为0.06-0.2%磷酸水溶液为流动相b,在体积比为5~95∶100~0的范围内梯度洗脱;
分别精密吸取供试品溶液和对照品溶液5~20μl,注入高效液相色谱仪,测定,分别得到供试品溶液和对照品溶液的液相色谱。
作为优选方案,所述步骤(3)包括设置hplc色谱条件:色谱柱采用十八烷基硅烷键合硅胶为填料,以waters2695acquityhplctm(4.6mm×250mm,5.0μm)为色谱柱,柱温30℃,流速为1.0ml/min,检测波长为230nm,并开启dad检测;以乙腈为流动相a,以体积分数为0.2%磷酸水溶液为流动相b,进行梯度洗脱,流动相a、b的比例变化为0~12min,a∶b为5%∶95%→11%∶89%;12~24min,a∶b为11%∶89%→11%∶89%;24~38min,a∶b为11%∶89%→23%∶77%;38~50min,a∶b为23%∶77%→28%∶72%;50~55min,a∶b为28%∶72%→31%∶69%;55~70min,a∶b为31%∶69%→100%∶0%;
分别精密吸取供试品溶液和对照品溶液10μl,注入高效液相色谱仪,测定,分别得到供试品溶液和对照品溶液的液相色谱。
在本发明的蓝芩口服液指纹图谱的建立方法的实施方案中,其中,所述步骤(4)采用的中药色谱指纹图谱相似度评价系统2004a版,所述步骤(4)包括:将步骤(3)得到的液相色谱的数据导入、多点校正、数据匹配,分析得到蓝芩口服液的指纹图谱。
在本发明的蓝芩口服液指纹图谱的建立方法的实施方案中,其中,所述步骤(4)得到的蓝芩口服液指纹图谱包括腺苷对应的1号峰、表告依春对应的4号峰、盐酸黄柏碱对应的5号峰、绿原酸对应的6号峰、木兰花碱对应的7号峰、栀子苷对应的9号峰、苯甲酸对应的15号峰、盐酸小檗碱对应的16号峰、黄芩苷对应的17号峰、汉黄芩苷对应的22号峰、黄芩素对应的24号峰、汉黄芩素对应的25号峰,其相对保留时间分别为0.120、0.587、0.711、0.777、0.806、1.000、1.882、1.946、2.002、2.615、2.856、3.003。
在本发明的蓝芩口服液指纹图谱的建立方法的实施方案中,其中,所述步骤(4)得到的蓝芩口服液指纹图谱的共有峰还包括相对保留时间为0.382的2号峰、相对保留时间为0.453的3号峰、相对保留时间为0.846的8号峰、相对保留时间为1.400的10号峰、相对保留时间为1.499的11号峰、相对保留时间为1.731的12号峰、相对保留时间为1.812的13号峰、相对保留时间为1.847的14号峰、相对保留时间为2.227的18号峰、相对保留时间为2.375的19号峰、相对保留时间为2.414的20号峰、相对保留时间为2.495的21号峰、相对保留时间为2.835的23号峰、相对保留时间为3.015的26号峰。
在本发明的蓝芩口服液指纹图谱的建立方法的实施方案中,其中,所述步骤(4)得到的蓝芩口服液指纹图谱如图2所示。
第二方面,本发明提供了上述蓝芩口服液指纹图谱的建立方法及其指纹图谱在建立其关键工艺中间体包括浓缩工艺中间体i、收膏工艺中间体ii、初配工艺中间体iii、精配工艺中间体iv的指纹图谱的应用,或者上述蓝芩口服液指纹图谱的建立方法及其指纹图谱在蓝芩口服液及其关键工艺中间体包括浓缩工艺中间体i、收膏工艺中间体ii、初配工艺中间体iii、精配工艺中间体iv中多个药材的多个药效成分进行定性定量分析的应用,或者上述蓝芩口服液指纹图谱的建立方法及其指纹图谱在制剂不同工艺环节的质量控制的应用。
本发明取得的技术效果至少包括下列一种或多种:
(1)以不同批次的蓝芩口服液得到的蓝芩口服液指纹图谱可以代表蓝芩口服液的大部分药效,更够有效的表征蓝芩口服液的质量。
(2)以蓝芩口服液的指纹图谱作为一个整体对待,注重各种化学成分的指纹特征峰及相互关系,注重蓝芩口服液的整体面貌特征,避免只测定一、二个指标性化学成分而判定蓝芩口服液质量的片面性,减少了人为主观判断而造成误差的可能性。
(3)以蓝芩口服液的指纹图谱为基础,建立了蓝芩口服液关键工艺中间体包括浓缩工艺中间体i、收膏工艺中间体ii、初配工艺中间体iii、精配工艺中间体iv的指纹图谱,并对各工艺步骤的多个药材的多个药效成分进行定性定量分析,根据含量变化的趋势和程度,确认关键工艺控制环节,制定相应的关键点质控标准,可用于蓝芩口服液的生产过程监控,对于全过程质量控制体系的建立提供了一定的理论依据和支持,保证临床用药的安全性和有效性。
(4)现有技术中,蓝芩口服液中单味药材的分离及含量测定多使用甲醇-水、乙腈-水、甲醇-0.2%磷酸水等进行梯度洗脱,本发明采用乙腈-0.2%磷酸水进行梯度洗脱。通过考察这四种系统的洗脱效果,比较色谱图(图3-图6),可见甲醇-水、甲醇-0.2%磷酸水系统洗脱的色谱图中,色谱峰的数量较少,不足以全面代表蓝芩口服液的物质基础;虽然乙腈-水系统洗脱的色谱图中色谱峰的数量与乙腈-0.2%磷酸水相近,但峰的分离度较差。本发明采用乙腈-0.2%磷酸水溶液作为洗脱溶剂,在该条件下得到的色谱峰显著地优于其他的洗脱系统。
本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过在说明书、权利要求书以及附图中所特别指出的结构来实现和获得。
附图说明
附图用来提供对本发明技术方案的进一步理解,并且构成说明书的一部分,与本申请的实施例一起用于解释本发明的技术方案,并不构成对本发明技术方案的限制。
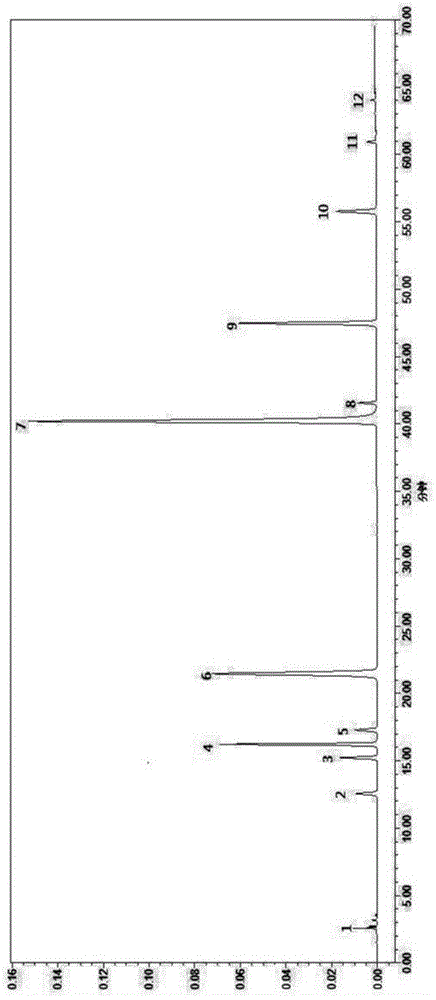
图1为12种对照品的hplc色谱图;
图1中,1:腺苷对照品,2:表告依春对照品,3:盐酸黄柏碱对照品,4:绿原酸对照品,5:木兰花碱对照品,6:栀子苷对照品,7:苯甲酸对照品,8:盐酸小檗碱对照品,9:黄芩苷对照品,10:汉黄芩苷对照品,11:黄芩素对照品,12:汉黄芩素对照品;
图2为本发明实施例1中19个批次蓝芩口服液指纹图谱叠加图谱;
图3为本发明实施例1中测得的蓝芩口服液的共有模式图谱;
图3中,1号峰:腺苷,4号峰:表告依春,5号峰:盐酸黄柏碱,6号峰:绿原酸,7号峰:木兰花碱,9号峰:栀子苷,15号峰:苯甲酸,16号峰:盐酸小檗碱,17号峰:黄芩苷,22号峰:汉黄芩苷,24号峰:黄芩素,25号峰:汉黄芩素;
图4为甲醇-0.2%磷酸水梯度洗脱测得的蓝芩口服液色谱图;
图5为甲醇-水梯度洗脱测得的蓝芩口服液色谱图;
图6为乙腈-水梯度洗脱测得的蓝芩口服液色谱图;
图7为本发明实施例1中板蓝根药材阳性对照色谱图;
图8为本发明实施例1中板蓝根药材阳性对照色谱图与指纹图谱的色谱对比图;
图9为本发明实施例1中板蓝根药材阴性对照色谱图;
图10为本发明实施例1中板蓝根药材阴性对照色谱图与指纹图谱的色谱对比图;
图11为本发明实施例1中黄芩药材阳性对照色谱图;
图12为本发明实施例1中黄芩药材阳性对照色谱图与指纹图谱的色谱对比图;
图13为本发明实施例1中黄芩药材阴性对照色谱图;
图14为本发明实施例1中黄芩药材阴性对照色谱图与指纹图谱的色谱对比图;
图15为本发明实施例1中栀子药材阳性对照色谱图;
图16为本发明实施例1中栀子药材阳性对照色谱图与指纹图谱的色谱对比图;
图17为本发明实施例1中栀子药材阴性对照色谱图;
图18为本发明实施例1中栀子药材阴性对照色谱图与指纹图谱的色谱对比图;
图19为本发明实施例1中黄柏药材阳性对照色谱图;
图20为本发明实施例1中黄柏药材阳性对照色谱图与指纹图谱的色谱对比图;
图21为本发明实施例1中黄柏药材阴性对照色谱图;
图22为本发明实施例1中黄柏药材阴性对照色谱图与指纹图谱的色谱对比图;
图23为本发明实施例3中9个批次蓝芩浓缩工艺中间体i指纹图谱叠加图谱;
图24为本发明实施例3中9个批次蓝芩浓缩工艺中间体i的共有模式图谱;
图25为本发明实施例3中9个批次蓝芩收膏工艺中间体ii指纹图谱叠加图谱;
图26为本发明实施例3中9个批次蓝芩收膏工艺中间体ii的共有模式图谱;
图27为本发明实施例3中6个批次蓝芩初配工艺中间体iii指纹图谱叠加图谱;
图28为本发明实施例3中6个批次蓝芩初配工艺中间体iii的共有模式图谱;
图29为本发明实施例3中6个批次蓝芩精配工艺中间体iv指纹图谱叠加图谱;
图30为本发明实施例3中6个批次蓝芩精配工艺中间体iv的共有模式图谱;
图31a-31c为本发明实施例4中蓝芩制备工艺过程中各指标成分的含量变化;
图32为本发明实施例4中蓝芩工艺中间体各指标成分的含量变化成分比例图。
具体实施方式
为使本申请的目的、技术方案和优点更加清楚明白,下文中将对本发明的实施例进行详细说明。需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互任意组合。
实施例用到的仪器及试剂如下:
1、仪器与试药
1.1仪器
十万分之一天平(sartoriusbt125d,赛多利斯科学仪器有限公司);台式高速离心机(tg16-ws,长沙湘仪离心机仪器有限公司);高效液相色谱仪(lc-10a,日本岛津公司,dad检测器);超声波清洗器(kh3200b,昆山禾创超声仪器有限公司);中药粉碎机(hc-150,黄城高速多功能粉碎机型号)。
1.2试药
腺苷对照品(批号:161207)、表告依春对照品(批号:170420)、盐酸黄柏碱对照品(批号:170307)、绿原酸对照品(批号:160507)、木兰花碱对照品(批号:170104)、栀子苷对照品(批号:170112)、苯甲酸对照品(批号:170507)、盐酸小檗碱对照品(批号:161223)、黄芩苷对照品(批号:170216)、汉黄芩苷对照品(批号:170109)、黄芩素(对照品批号:170324)、汉黄芩素对照品(批号:170320),以上标准品均购自上海融禾医药科技有限公司,含量≥98%。
色谱甲醇、乙腈购自美国sigma公司,色谱磷酸购于天津科密欧化学试剂有限公司。所用水为屈臣氏蒸馏水。
共收集19个批次的蓝芩口服液样品,均由扬子江药业集团有限公司生产。19个批次的蓝芩口服液供试样本基本信息见表1。
表1蓝芩口服液供试样本基本信息
共收集9个批次的蓝芩浓缩工艺中间体i样品,均由扬子江药业集团有限公司生产。9个批次的蓝芩浓缩工艺中间体i供试样本基本信息见表2。
表2蓝芩浓缩工艺中间体i供试样本基本信息
共收集9个批次的蓝芩收膏工艺中间体ii样品,均由扬子江药业集团有限公司生产。9个批次的蓝芩收膏工艺中间体ii供试样本基本信息见表3。
表3蓝芩收膏工艺中间体ii供试样本基本信息
共收集6个批次的蓝芩初配工艺中间体iii样品,均由扬子江药业集团有限公司生产。6个批次的蓝芩初配工艺中间体iii供试样本基本信息见表4。
表4蓝芩初配工艺中间体iii供试样本基本信息
共收集6个批次的蓝芩精配工艺中间体iv样品,均由扬子江药业集团有限公司生产。6个批次的蓝芩初配工艺中间体iv供试样本基本信息见表5。
表5蓝芩初配工艺中间体iv供试样本基本信息
实施例1蓝芩口服液指纹图谱的建立方法及方法学考察
1.蓝芩口服液指纹图谱的建立方法
蓝芩口服液指纹图谱的建立方法,包括以下步骤:
(1)供试品溶液的制备:取以上表1的19个批次不同的蓝芩口服液,精密吸取200μl于10ml离心管中,加入4.8ml的甲醇进行稀释,混匀后过0.45μm微孔滤膜,作为供试品溶液;
(2)对照品溶液的制备:取黄芩苷对照品适量,精密称定,加70%甲醇溶解,配制成含量为208μg·ml-1的对照品溶液,分别取腺苷对照品、表告依春对照品、盐酸黄柏碱对照品、绿原酸对照品、木兰花碱对照品、栀子苷对照品、苯甲酸对照品、盐酸小檗碱对照品、汉黄芩苷对照品、黄芩素对照品、汉黄芩素对照品适量,精密称定,加甲醇溶解,配制成含量分别为7.52μg·ml-1、3.84μg·ml-1、6.44μg·ml-1、32.96μg·ml-1、2.52μg·ml-1、284.00μg·ml-1、162.00μg·ml-1、4.56μg·ml-1、98.00μg·ml-1、10.60μg·ml-1、9.76μg·ml-1的对照品溶液。
(3)hplc色谱条件:色谱柱采用十八烷基硅烷键合硅胶为填料,以waters2695acquityhplctm(4.6mm×250mm,5.0μm)为色谱柱,柱温30℃,流速为1.0ml/min,检测波长为230nm,并开启dad检测。以乙腈为流动相a,以体积分数为0.2%磷酸水溶液为流动相b,进行梯度洗脱,流动相a、b的比例变化为0~12min,a∶b为5%∶95%→11%∶89%;12~24min,a∶b为11%∶89%→11%∶89%;24~38min,a∶b为11%∶89%→23%∶77%;38~50min,a∶b为23%∶77%→28%∶72%;50~55min,a∶b为28%∶72%→31%∶69%;55~70min,a∶b为31%∶69%→100%∶0%;
(4)分别精密吸取供试品溶液和对照品溶液10μl,注入高效液相色谱仪,测定,分别得到供试品溶液和对照品溶液的液相色谱。
(5)将液相图谱导入指纹图谱相似度评价软件(中药色谱指纹图谱相似度评价系统2004a版)分析,以17050221批次口服液作为参照图谱,选取“时间窗宽度”为0.1min,采用平均数法计算,多点校正、数据匹配,生成叠加色谱图(见图3)和对照图谱(见图2)。将19批蓝芩口服液的指纹图谱与生成的对照指纹图谱进行相似度分析,结果见表6。
表619批蓝芩口服液指纹图谱相似度
由表6可知,不同批次蓝芩口服液指纹图谱与对照图谱的相似度均在0.995~1之间,表明不同批次间的相似度较高,工艺相对稳定。
经与12种对照品比对,确认2.58min峰为腺苷(1号峰)、12.56min峰为表告依春(4号峰),15.18min峰为盐酸黄柏碱(5号峰),16.68min峰为绿原酸(6号峰),17.23min峰为木兰花碱(7号峰),21.53min峰为栀子苷(9号峰),40.13min峰为苯甲酸(15号峰),41.45min峰为盐酸小檗碱(16号峰),47.43min峰为黄芩苷(17号峰),55.69min峰为汉黄芩苷(22号峰),60.88min峰为黄芩素(24号峰),64.01min峰为汉黄芩素(25号峰)。其中选取分离度较好,峰面积较大且稳定,保留时间适中的栀子苷为参照峰(s)。以栀子苷为参照峰的hplc指纹图谱共确定26个共有峰。
2.指纹图谱方法学考察
2.1精密度试验
取蓝芩口服液(17050221),按实施例1的供试品溶液的制备方法制备供试品溶液,按实施例1中色谱条件连续进样6针,得到6张色谱图。以栀子苷峰为参照峰,计算各共有峰与参照峰的相对峰面积及相对保留时间,并计算rsd值,结果见表7和表8。
表7蓝芩口服液指纹图谱精密度试验考察结果(各共有峰相对峰面积)
表8蓝芩口服液指纹图谱精密度试验考察结果(各共有峰相对保留时间)
由表7和表8可知,各共有色谱峰的相对峰面积rsd%<3%、相对保留时间rsd%<3%,说明该方法精密度良好。
2.2重复性试验
取蓝芩口服液(17050221),平行称取6份,按实施例1的供试品溶液的制备方法制备6份供试品溶液,按实施例1中色谱条件分别进样,得到6张色谱图。以栀子苷峰为参照峰,计算各共有峰与参照峰的相对峰面积及相对保留时间,并计算rsd值,结果见表9和表10。
表9蓝芩口服液指纹图谱重复性试验考察结果(各共有峰相对峰面积)
表10蓝芩口服液指纹图谱重复性试验考察结果(各共有峰相对保留时间)
由表9和表10可知,各共有色谱峰的相对峰面积rsd%<3%、相对保留时间rsd%<3%,说明该方法重复性良好。
2.3稳定性试验
取蓝芩口服液(17050221),按实施例1的供试品溶液的制备方法制备6份供试品溶液,按实施例1中色谱条件,分别于0h、2h、4h、8h、12h、24h条件进样,得到6张色谱图。以黄芩苷峰为参照峰,计算各共有峰与参照峰的相对峰面积及相对保留时间,并计算rsd值,结果见表11和表12。
表11蓝芩口服液指纹图谱稳定性试验考察结果(各共有峰相对峰面积)
表12蓝芩口服液指纹图谱稳定性试验考察结果(各共有峰相对保留时间)
由表11和表12可知,各共有色谱峰的相对峰面积rsd%<3%、相对保留时间rsd%<3%,说明供试品溶液在24h内基本稳定。
3.蓝芩口服液与各原料药材相关性及共有峰归属分析
按实施例1方法测定蓝芩口服液中板蓝根、黄芪、栀子、黄柏的阳性对照色谱图及阴性对照色谱图,通过dad检测器对蓝芩口服液指纹图谱与各药材阳性对照色谱图及阴性对照色谱图中色谱峰的紫外吸收进行分析,比较色谱峰的保留时间,最终确认蓝芩口服液指纹图谱中共有色谱峰在药材图谱上的归属峰。
3.1板蓝根中的共有峰归属分析
板蓝根药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图,见图4和图5。板蓝根药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图,见图6和图7。通过色谱图对比,可知指纹图谱共有峰中的1,4号峰来自板蓝根药材。
3.2黄芩中的共有峰归属分析
黄芩药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图,见图8和图9。黄芩药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图,见图10和图11。通过色谱图对比,可知指纹图谱共有峰中的12,13,14,18,19,20,21,22,23,24,25,26号峰来自黄芩药材。
3.3栀子中的共有峰归属分析
栀子药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图,见图12和图13。栀子药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图,见图14和图15。通过色谱图对比,可知指纹图谱共有峰中的2,3,6,9,13,14,15,17号峰来自栀子药材。
3.4黄柏中的共有峰归属分析
黄柏药材的阳性对照色谱图及阳性对照色谱图与指纹图谱的色谱对比图,见图16和图17。黄柏药材的阴性对照色谱图及阴性对照色谱图与指纹图谱的色谱对比图,见图18和图19。通过色谱图对比,可知指纹图谱共有峰中的5,7,8,10,11,16号峰来自黄柏药材。
3.5蓝芩口服液与各药材共有峰归属分析
由上述结果分析可知,蓝芩口服液指纹图谱中的26个共有峰,在处方中板蓝根、黄芩、栀子、黄柏4味药材阳性对照与阴性对照色谱图中,基本找到明确归属。可知指纹图谱共有峰中的1,4号峰来自板蓝根药材;共有峰中的12,18,19,20,21,22,23,24,25,26号峰来自黄芩药材;共有峰中的5,7,8,10,11,16号峰来自黄柏药材;共有峰中的2,3,6,9,15,17号峰来自栀子药材;13号与14号峰同时存在于黄芩和栀子药材中。由此可见,蓝芩口服液的指纹图谱基本能代表蓝芩口服液的物质基础。
实施例2蓝芩口服液指纹图谱的多波长定量成分的检测及方法学考察
1.多波长定量成分的检测
腺苷、表告依春、盐酸黄柏碱、绿原酸、木兰花碱、栀子苷、苯甲酸、盐酸小檗碱、汉黄芩苷、黄芩素、汉黄芩素12种药效指标成分,保留时间分别为:2.58,12.56,15.18,16.68,17.23,21.53,40.13,41.45,47.43,55.69,60.88,64.01min。开启dad检测器进行全波长扫描,比较不同波长条件下各药效指标成分的峰面积,得到各指标成分的最适检测波长,结果显示,腺苷、表告依春、盐酸黄柏碱、绿原酸、木兰花碱、栀子苷、苯甲酸和盐酸小檗碱在230nm条件下有较大吸收;黄芩苷、汉黄芩苷、黄芩素、汉黄芩素在276nm条件下有较大吸收。最终选取在230nm条件下记录腺苷、表告依春、盐酸黄柏碱、绿原酸、木兰花碱、栀子苷、苯甲酸、盐酸小檗碱的保留时间和峰面积,计算各特征峰的rsd值;在276nm条件下记录黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的保留时间和峰面积,计算各特征峰的rsd值。
2.多波长定量成分检测方法学考察
2.1线性考察
精密量取黄芩苷对照品溶液适量,加70%的甲醇逐级稀释,配置为一系列浓度梯度的对照品溶液,在所建立的色谱条件下进样检测,于波长276nm下测定并记录峰面积;精密量取混合对照品溶液适量,加甲醇逐级稀释,配置为一系列浓度梯度的混合对照品溶液,在所建立的色谱条件下进样检测,于波长230nm和276nm下测定并记录峰面积;以峰面积为纵坐标(y),浓度(x)为横坐标,求得各指标成分的回归方程如表13所示:
表13各药效指标成分的回归方程
2.2精密度试验
取蓝芩口服液(17050232),按实施例1的供试品溶液的制备方法制备供试品溶液,按实施例1中色谱条件连续进样6针,得到6张色谱图。计算各特征峰的峰面积及保留时间,代入回归方程计算各指标成分的含量,计算rsd值,结果见表14和表15。
表14各特征峰精密度试验的保留时间
表15各特征峰精密度试验的含量(μg·ml-1)
由表14和表15可知,各特征峰保留时间的rsd值均<3%,含量的rsd值均<3%,说明该方法精密度良好。
2.3重复性试验
取蓝芩口服液(17050232),按实施例1的供试品溶液的制备方法,平行制备6份供试品溶液,按实施例1中色谱条件分别进样,得到6张色谱图。计算各特征峰的峰面积及保留时间,代入回归方程计算各指标成分的含量,计算rsd值,结果见表16和表17。
表16各特征峰重复性试验的保留时间
表17各特征峰重复性试验的含量(μg·ml-1)
由表16和表17可知,各特征峰保留时间的rsd值均<3%,含量的rsd值均<3%,说明该方法重复性良好。
2.4稳定性试验
取蓝芩口服液(17050232),按实施例1的供试品溶液的制备方法,平行制备6份供试品溶液,按实施例1中色谱条件,分别于0h、2h、4h、8h、12h、24h条件进样,得到6张色谱图。计算各共特征峰的峰面积及保留时间,代入回归方程计算各指标成分的含量,结果见表18和表19。
表18各特征峰稳定性试验的保留时间
表19各特征峰稳定性试验的含量(μg·ml-1)
由表18和表19可知,各特征峰保留时间的rsd值均<3%,含量的rsd值<3%,说明特征成分在室温条件下24h内稳定性良好。
2.5加样稳定性试验
称取同一批次蓝芩口服液,按实施例1的供试品溶液的制备方法制备9份供试品溶液,分别精密加入含腺苷、表告依春、盐酸黄柏碱、绿原酸、木兰花碱、栀子苷、苯甲酸、盐酸小檗碱、汉黄芩苷、黄芩素、汉黄芩素对照品溶液适量,按实施例1中色谱条件,分别进样,得到9张色谱图。计算各指标化学成分的平均加样回收率及rsd值。结果见表20-表31。
表20腺苷的加样回收率结果
由表20可知,腺苷的加样回收率结果符合试验要求。
表21表告依春的加样回收率结果
由表21可知,表告依春的加样回收率结果符合试验要求。
表22盐酸黄柏碱的加样回收率结果
由表22可知,盐酸黄柏碱的加样回收率结果符合试验要求
表23绿原酸的加样回收率结果
由表23可知,绿原酸的加样回收率结果符合试验要求。
表24木兰花碱的加样回收率结果
由表24可知,木兰花碱的加样回收率结果符合试验要求。
表25栀子苷的加样回收率结果
由表25可知,栀子苷的加样回收率结果符合试验要求。
表26苯甲酸的加样回收率结果
由表26可知,苯甲酸的加样回收率结果符合试验要求。
表27盐酸小檗碱的加样回收率结果
由表27可知,盐酸小檗碱的加样回收率结果符合试验要求。
表28黄芩苷的加样回收率结果
由表28可知,黄芩苷的加样回收率结果符合试验要求。
表29汉黄芩苷的加样回收率结果
由表29可知,汉黄芩苷的加样回收率结果符合试验要求。
表30黄芩素的加样回收率结果
由表30可知,黄芩素的加样回收率结果符合试验要求。
表31汉黄芩素的加样回收率结果
由表31可知,汉黄芩素的加样回收率结果符合试验要求。
3、指标成分含量的测定
取以上表1的19个不同批次蓝芩口服液,按实施例1的供试品溶液的制备方法制备19份供试品溶液,按实施例1中色谱条件分别进样,记录各指标成分的峰面积,代入线性回归方程计算含量,每个样品重复测量两次,取平均值。计算不同批次蓝芩口服液中各指标化学成分含量。结果见表32。
由表32可知,蓝芩口服液成品制剂不同批次间12种药效指标成分除部分指标成分外,不同批次间指标成分含量差别不显著。证明蓝芩口服液质量均一稳定。
实施例3蓝芩口服液关键工艺中间体指纹图谱的建立
1.浓缩工艺中间体i的指纹图谱
取以上表2的9个不同批次的蓝芩浓缩工艺中间体i适量,加60%甲醇水溶液稀释,混匀,过0.45μm微孔滤膜,作为供试品溶液。按实施例1中色谱条件进样,得到供试品溶液的液相色谱。将液相图谱导入国家药典委员会编写的“色谱指纹图谱相似度评价系统2004a版”中对色谱图进行数据处理。得到浓缩工艺中间体i的叠加色谱图(见图20)及对照图谱(见图21),将9批蓝芩浓缩工艺中间体i的指纹图谱与生成的对照指纹图谱进行相似度分析,结果见表33。
表339批蓝芩浓缩工艺中间体i指纹图谱相似度
由表33可知,不同批次浓缩中间体i指纹图谱与对照图谱的相似度均在0.990~1之间,表明不同批次间的相似度较高,工艺相对稳定。
2.收膏工艺中间体ii的指纹图谱
取以上表3的9个不同批次的蓝芩收膏工艺中间体ii适量,加60%甲醇水溶液稀释,混匀,过0.45μm微孔滤膜,作为供试品溶液。按实施例1中色谱条件进样,得到供试品溶液的液相色谱。将液相图谱导入国家药典委员会编写的“色谱指纹图谱相似度评价系统2004a版”中对色谱图进行数据处理。得到收膏工艺中间体ii的叠加色谱图(见图22)及对照图谱(见图23),将9批蓝芩收膏工艺中间体ii的指纹图谱与生成的对照指纹图谱进行相似度分析,结果见表34。
表349批蓝芩收膏工艺中间体ii指纹图谱相似度
由表34可知,不同批次收膏工艺中间体ii指纹图谱与对照图谱的相似度均在0.992~1之间,表明不同批次间的相似度较高,工艺相对稳定。
3.初配工艺中间体iii的指纹图谱
取以上表4的6个不同批次的蓝芩初配工艺中间体iii适量,加60%甲醇溶液稀释,混匀,过0.45μm微孔滤膜,作为供试品溶液。按实施例1中色谱条件进样,得到供试品溶液的液相色谱。将液相图谱导入国家药典委员会编写的“色谱指纹图谱相似度评价系统2004a版”中对色谱图进行数据处理。得到初配工艺中间体iii的叠加色谱图(见图24)及对照图谱(见图25),将6批蓝芩初配工艺中间体iii的指纹图谱与生成的对照指纹图谱进行相似度分析,结果见表35。
表356批蓝芩初配工艺中间体iii指纹图谱相似度
由表35可知,不同批次初配工艺中间体iii指纹图谱与对照图谱的相似度均在0.999~1之间,表明不同批次间的相似度较高,工艺相对稳定。
4.精配工艺中间体iv的指纹图谱
取以上表5的6个不同批次的蓝芩精配工艺中间iv适量,加60%甲醇水溶液稀释,混匀,过0.45μm微孔滤膜,作为供试品溶液。按实施例1中色谱条件进样,得到供试品溶液的液相色谱。将液相图谱导入国家药典委员会编写的“色谱指纹图谱相似度评价系统2004a版”中对色谱图进行数据处理。得到精配工艺中间体iv的叠加色谱图(见图26)及对照图谱(见图27),将6批蓝芩精配工艺中间体iv的指纹图谱与生成的对照指纹图谱进行相似度分析,结果见表36。
表376批蓝芩精配工艺中间体iv指纹图谱相似度
由表36可知,不同批次精配工艺中间体iv指纹图谱与对照图谱的相似度均在0.999~1之间,表明不同批次间的相似度较高,工艺相对稳定。
实施例4蓝芩口服液关键工艺中间体药效指标成分的含量变化
取蓝芩口服液各关键工艺中间体适量,制成供试品溶液,按实施例1中色谱条件进样检测,于波长230nm和276nm下测定并记录各药效指标成分在制剂生产各关键工艺中间体的峰面积,结果见表37。考察各药效成分的含量变化趋势及程度(见图28-图29)。
表3712种药效指标成分在制备工艺过程中的峰面积变化
由结果可知,不同工艺过程各成分的变化规律如下:由浓缩过程到收膏过程,除腺苷含量有轻微的升高外,其余各成分均出现显著的含量下降现象;由收膏过程到初配环节,表告依春含量急剧下降,盐酸黄柏碱、绿原酸、栀子苷、腺苷、汉黄芩素含量升高,木兰花碱、黄芩苷、汉黄芩苷、黄芩素含量轻微升高,苯甲酸含量急剧大幅度升高,盐酸小檗碱含量有轻微下降的趋势;由初配环节到精配环节,12种药效指标成分的含量均有不同程度的下降趋势;由精配环节到制剂终产品,表告依春含量急剧大幅度增加,木兰花碱含量升高;盐酸黄柏碱、绿原酸、栀子苷、苯甲酸、盐酸小檗碱、腺苷、黄芩苷、汉黄芩苷、黄芩素、汉黄芩素的含量均有不同程度的下降。以单个药效指标成分为依据,其含量受工艺影响的变化规律如下:表告依春在浓缩、收膏、初配这三个环节,其含量呈大幅度急剧下降趋势,精配液含量变化不大,由精配到终产品含量又呈现急剧上升至略高于浓缩液的含量,可知收膏步骤、初配环节及终产品环节为表告依春含量控制的关键工艺步骤。盐酸黄柏碱由浓缩过程到收膏过程含量明显下降,其他工艺环节含量变化不大,故应在收膏过程对盐酸黄柏碱的含量加以控制。绿原酸除在初配环节含量小幅度增加外,其余的工艺环节均呈下降趋势;木兰花碱、栀子苷、盐酸小檗碱、黄芩苷、汉黄芩苷、黄芩素、汉黄芩素在醇沉收膏环节含量下降趋势明显,应为关键控制步骤,其他工艺环节相对含量的变化不大;苯甲酸在浓缩液与收膏中含量均比较低,在初配环节由于添加剂苯甲酸钠的加入含量急剧上升,之后工艺过程中其含量稍微降低;腺苷在初配环节含量达到最大值,精配液和终产品中其含量略微降低,与浓缩液与收膏中的含量差别不大。
以上试验结果表明,本发明提供的一种蓝芩口服液指纹图谱的建立方法及其指纹图谱,较全面分析了蓝芩口服液的整体成分,能够基本代表反映蓝芩口服液的有效成分和物质基础,可以通过建立蓝芩口服液及其工艺中间体的指纹图谱,对口服液各指标成分在不同工艺过程含量及其变化规律进行检测,对制剂不同工艺环节的质量控制有一定的鉴别意义,对于全过程质量控制体系的建立提供了一定的理论依据和支持,能较全面的反映蓝芩口服液的质量信息,保证产品质量均一稳定。研究发现:1、蓝芩口服液指纹图谱中的26个共有峰,在处方的板蓝根、黄芩、栀子、黄柏4味药材阳性对照与阴性对照色谱图中,基本找到明确归属。可知指纹图谱共有峰中的1,4号峰来自板蓝根药材;共有峰中的12,18,19,20,21,22,23,24,25,26号峰来自黄芩药材;共有峰中的5,7,8,10,11,16号峰来自黄柏药材;共有峰中的2,3,6,9,15,17号峰来自栀子药材;13号与14号峰同时存在于黄芩和栀子药材中。由此可见,蓝芩口服液的指纹图谱基本能代表蓝芩口服液的物质基础。2、12种药效指标成分在各个工艺中间体中的含量相较于蓝芩口服液有不同的变化趋势和变化程度,但均具有较明显的含量变化,故应根据具体变化制定相应的关键点质控标准。浓缩液及初配液中大部分指标成分的含量显著性增加,收膏液及精配液中大部分指标成分的含量无显著性差异,故浓缩环节及初配环节为关键工艺控制环节,应建立关键点质控标准,确保从中间体环节控制中药制剂的质量。
虽然本申请所揭露的实施方式如上,但所述的内容仅为便于理解本申请而采用的实施方式,并非用以限定本申请。任何本申请所属领域内的技术人员,在不脱离本申请所揭露的精神和范围的前提下,可以在实施的形式及细节上进行任何的修改与变化,但本申请的专利保护范围,仍须以所附的权利要求书所界定的范围为准。
- 还没有人留言评论。精彩留言会获得点赞!