一种氨基化合物的衍生化方法及其应用
本发明属于分析化学技术领域,具体涉及一种氨基化合物的衍生化方法及其应用。
背景技术:
神经递质(nts)是内源性化学信使,能够促进神经传递并在许多大脑功能(包括行为和认知)中发挥重要作用。它们影响并调节肌张力和心率,调节学习,睡眠,记忆,意识,情绪和食欲。在正常的生理条件下,神经递质在控制突触功能的细胞中形成神经网络。神经递质通常存储在突触小泡中,并通过适当的信号释放到突触中。释放的神经递质通过突触间隙建立连接,并与相应的受体结合以完成突触传递。中枢神经系统中神经递质浓度的变化与许多精神和身体疾病有关,例如阿尔茨海默病(ad),帕金森氏病,精神分裂症,癫痫病,抑郁症等。因此,神经递质浓度的检测是研究这些疾病的重要手段。有效监测体内神经递质浓度对于疾病的诊断和治疗以及新药的研发至关重要。
针对神经递质检测已报道的方法可总结为仪器分析方法、电化学传感器、新兴材料检测法3大类。在神经递质检测中使用较为广泛的仪器分析法,主要包括高效液相色谱(hplc)法、高效毛细管电泳(hpce)法、气相色谱-质谱联用(gc-ms)法以及液相色谱-质谱联用(lc-ms)法等。但是,生物样品本身由复杂的基质和许多内源性干扰物质组成,大多数神经递质是极性物质,它们在反相色谱柱上的保留能力极弱,并且具有不理想的电离效率。因此,分析和检测神经递质极为困难。
蛋白质直接参与机体的生理/病理变化,并且是了解与细胞功能相关的基本机制的主要载体。蛋白质的鉴定可以提供有关其相互作用和翻译后修饰的信息,这有助于了解它们的生物学功能或疾病状态。最广泛使用的蛋白质鉴定策略是自下而上的蛋白质组学,它是基于从目标细胞或组织蛋白质混合物中获得的蛋白水解肽的鉴定,然后进行液相色谱-串联质谱(lc-ms/ms)分析。
多肽的分析检测中,ms信号可能由于其组成差异而有很大差异,主要是由于两个原因:(1)差的电离效率;(2)反相色谱中的分离度受限。
目前还没有能够同时应有与以上物质的检测,且能够减少杂质的干扰,改善被分析物的信号强度的分析方法。
技术实现要素:
为解决上述技术问题,本申请提供一种氨基化合物的衍生化方法及其应用,所述方法通过采用吡喃盐对待测样品进行衍生化处理,降低了样品基质、无机盐及内源性杂质带来的干扰,便于色谱分离和质谱检测。
为实现上述目的,本发明采用的技术方案如下:
本申请一方面,提供了一种氨基化合物的衍生化方法,至少包括:
将含有氨基化合物、衍生化试剂的混合物进行衍生化反应,得到衍生化产物;
衍生化试剂为吡喃盐。
可选地,吡喃盐为α-活性吡喃盐。
α-活性吡喃盐是指吡喃盐上α位没有取代基或只有一个取代基。
可选地,吡喃盐上α位无取代基或吡喃盐上有至少一个取代基;
所述取代基包括c6~c10的芳基、c1~c6的烷基中的至少一种。
芳基为苯基或苯基取代基中的至少一种,苯基取代基包括对甲基苯基、甲氧基取代苯基。
烷基为甲基、乙基、丙基、丁基、戊基、己基中的至少一种。
具体地,吡喃盐可以为2,4,6-三苯基吡喃鎓四氟硼酸盐、2,4,6-三甲基吡喃鎓四氟硼酸盐、2,3,4,5-四苯基吡喃鎓四氟硼酸盐、2,6-二甲基-4-苯基吡喃鎓四氟硼酸盐等。
吡喃盐可与单胺类神经递质反应生成单种产物,该产物是与单胺类神经递质中的氨基发生氮氧交换反应的产物,经实验验证,衍生盐过量添加不会对检测结果产生不良影响。
吡喃盐具有永久性正电荷和多个疏水基团(苯基,烷基)的结构,通过吡喃盐衍生化标记,在多肽中引入待正电荷的基团,同时增强其疏水性,有利于提高质谱分析检测中离子的电离产率,增强信号强度。另外永久正电荷标记改善了多肽cid碎裂模式,有助于多肽/蛋白的从头测序。
可选地,待测样品与衍生化试剂的摩尔比为1:1~1000。
具体地,待测样品与衍生化试剂的摩尔比可独立选自1:1、1:10、1:20、1:50、1:100、1:1000。
可选地,衍生化反应的条件包括:反应温度为20~90℃;反应时间为0.5~4h。
具体地,反应温度的下限独立选自20℃、30℃、40℃、50℃、60℃;反应温度的上限独立选自35℃、55℃、65℃、70℃、90℃。
反应时间独立选自0.5h、1h、1.5h、2h、3h、3.5h、4h。
可选地,催化剂在混合物中的浓度为100μmol/l~100mmol/l;
催化剂为三乙胺、三乙胺乙酸缓冲液中的至少一种;
优选地,teaa缓冲液ph为6~11。
teaa缓冲液是指三乙胺-醋酸混合溶液,通过对其ph的限定,控制二者的混合比例。
具体地,三乙胺或teaa缓冲液作为催化剂,能够促进吡喃盐与氨基化合物或氨基化合物标准物质的反应。为促使氨基化合物或氨基化合物标准物质、吡喃盐在溶剂中分散、溶解完全,可先将氨基化合物或氨基化合物标准物质、吡喃盐溶于少量溶剂中,再向溶剂中加入三乙胺或teaa缓冲液。
具体地,催化剂在混合物中的浓度下限可独立选自100μmol/l、500μmol/l、1mmol/l、10mmol/l、20mmol/l;催化剂在混合物中的浓度上限可独立选自20mmol/l、40mmol/l、60mmol/l、80mmol/l、100mmol/l。
可选地,混合物中还包括溶剂;
待测样品与溶剂的摩尔体积比为0.5nmol/l~1mmol/l;
溶剂选自水、乙腈、乙醇、甲醇、dmso、二甲基甲酰胺、四氢呋喃中的至少一种。
具体地,氨基化合物与溶剂的摩尔体积比的下限可独立选自0.5nmol/l、50nmol/l、100nmol/l、500nmol/l、1000nmol/l;氨基化合物与溶剂的摩尔体积比的上限可独立选自10μmol/l、100nmol/l、500nmol/l、800nmol/l、1mmol/l。
本申请实施例中优选采用乙腈。
可选地,氨基化合物为单胺类神经递质、多肽、氨基酸中的至少一种。
具体地,单胺类神经递质为人或动物体内各种体液提取物中的伯胺类神经递质,如多巴胺、血清素、4-氨基丁酸等。
可选地,单胺类神经递质选自多巴胺(da)、组胺(histamine)、5-羟色胺(5-ht)、去甲肾上腺素(ne)、4-氨基丁酸(gaba)中的至少一种。
本申请又一方面,提供了一种氨基化合物的定性/定量分析方法,所述分析方法至少包括以下步骤:
步骤1、对氨基化合物进行衍生化处理,得到衍生化产物;
所述衍生化处理方法为上述任一衍生化方法;
步骤2、对所述衍生化产物进行液相色谱-质谱分析。
可选地,液相色谱-质谱分析还包括:
将液相色谱-质谱分析结果与标准曲线对照,获得氨基化合物的含量;
优选地,标准曲线的获得方法包括:
将氨基化合物的标准物质加入溶剂b,配制成系列浓度的标准溶液;
将含有衍生化试剂的原料加入到含有氨基化合物标准物质的溶液中进行衍生化反应,得到衍生化产物;
对衍生化产物进行液相色谱-质谱分析,得到衍生化产物所对应的峰面积;
对衍生化产物所对应的峰面积与标准溶液浓度作回归分析,得到物质浓度与峰面积的关系曲线。
可选地,氨基化合物的标准物质与衍生化试剂的摩尔比至少为1:1。
具体地,氨基化合物的标准物质与衍生化试剂的摩尔比可独立选自1:1、1:10、1:20、1:50、1:100。
可选地,溶剂b选自乙腈、乙醇、甲醇、dmso、二甲基甲酰胺、四氢呋喃中的至少一种;
优选地,液相色谱条件为:
流动相a为体积百分数0.1%的甲酸,流动相b为体积百分数为100%乙腈;
洗脱梯度:0~10min,5~100%b;10~13min,100%b;13~13.1min,100~5%b;13.1~16min,5%b;
优选地,所述质谱分析采用电喷雾电离源正离子模式;
进一步优选地,所述质谱分析为二级质谱分析;
更进一步优选地,所述二级质谱分析中,cid模式能量范围为10~40ev。
具体地,cid模式能量可独立选自10ev、20ev、30ev、40ev,或者上述两个点值之间的任一点值。
本申请又一方面,提供了上述任一氨基化合物的衍生化方法、任一氨基化合物的定性/定量分析方法在多肽或/和蛋白质测序中的应用。
本发明的有益效果在于:
1、本发明通过化学修饰技术对氨基化合物进行衍生化处理,改变其物理化学特性,从而提高液相色谱-质谱联用检测中的离子化效率,降低基质干扰;同时减少无机盐及内源性杂质的干扰,改善被分析物的信号强度,便于色谱分离和质谱检测,使得采用液相色谱-质谱联用技术分析对衍生化产物检测,能实现对氨基化合物的定性、定量分析。
2、相对于现有的氨基化合物检测技术,本发明方法操作简单、成本低,采用常规色谱柱即可实现目标物与杂质的有效分离,且质谱分析效果良好,检测灵敏度高,准确性好。
3、本发明采用吡喃盐作为化学衍生化试剂,其具有荧光性能良好、衍生效率高、廉价易得等优点。
4、本申请衍生化采用吡喃盐标记,可以带来多肽n端永久正电荷,从而明显增强n端cid碎片离子,且对于蛋白质的信号增强同样有效,能够简化多肽/蛋白的测序分析,具有很高的应用前景。
4、本发明质谱分析时采用电喷雾电离源正离子模式能显著提高检测的灵敏度,适用于低含量氨基化合物的分析检测。
附图说明
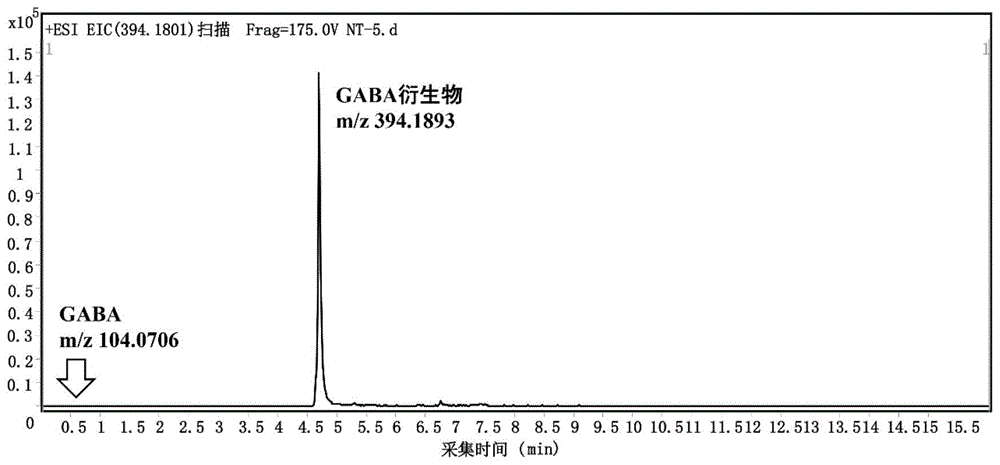
图1为本发明实施例1中gaba衍生化产物的提取离子色谱图;
图2为本发明实施例1中gaba衍生化产物的质谱同位素分布图;
图3为本发明实施例1中色谱提取结果对比图,其中图a为2,4,5-三苯基吡喃鎓四氟硼酸盐色谱提取结果,图b为gaba衍生化产物对应色谱提取结果;
图4为本发明实施例1中gaba的衍生化产物在0.5~1000nm浓度范围内的标准曲线;
图5为本发明实施例2中五种单胺类神经递质混合衍生化产物的提取离子色谱图,图a~e分别为gaba、组胺、多巴胺、去甲肾上腺素和5-羟色胺的衍生化标记结果;
图6为本发明实施例2中十种吡喃盐与五种单胺类神经递质的增敏能力对比图;
图7为实施例4中多肽标记的两种主要形式示意图;
图8为实施例4中五种多肽衍生化前后质谱信号的变化结果;
图9为实施例5中多肽hgtvvl的cid裂解结果;
图10为实施例6中肌红蛋白和细胞色素c酶解产物衍生化前后色谱结果,其中图a和图c为肌红蛋白和细胞色素c酶解产物衍生化前的色谱结果,其中图b和图d为肌红蛋白和细胞色素c酶解产物衍生化后的色谱结果;
图11为细胞色素c酶解多肽片段的ms/ms分析结果,其中(a),(b)和(c)分别为多肽lenpk的三种衍生化产物p6-lenpk,p5-lenpk-p5和p5-lenpk,这三种衍生物的离子类型分别为[m+h+p6]2+,[m+2p5]2+和[m+h+p5]2+;(d),(e)和(f)分别为以上三种衍生化产物的esi-ms/ms光谱以及相应的cid碎片。
具体实施方式
下面结合附图和具体实施例,进一步阐述本发明。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法;下述实施例中所用的试剂、材料等,如无特殊说明,均可从商业途径得到。
实施例1
基于柱前衍生的lc-ms测定小鼠大脑中4-氨基丁酸(gaba)含量的方法,包括以下步骤:
1)吡喃盐衍生化试剂、三乙胺溶液及gaba标准溶液的配制:
准确称取3.95mg的2,4,5-三苯基吡喃鎓四氟硼酸盐于干燥的试剂小瓶中,加入10ml无水乙腈配制成浓度1mmol/l的储备溶液a,密封后置于冰箱中储存备用;
准确称取1.03mg的gaba于干燥的试剂小瓶中,加入10ml无水乙醇配制成浓度1mmol/l的储备溶液b,密封后置于冰箱中储存备用;
准确量取0.139ml的三乙胺加入9.861ml乙腈溶解,配制成浓度100mmol/l的储备溶液c,密封后置于冰箱中储存备用;
取10个干燥的离心管,分别量取储备溶液c配制成浓度为1μmol/l、0.5μmol/l、0.1μmol/l、50nmol/l、20nmol/l、10nmol/l、5nmol/l、1nmol/l、0.8nmol/l、0.5nmol/l的系列gaba标准溶液;
2)制作标准曲线
衍生化反应:分别量取100μl系列浓度的gaba标准溶液于1.5ml的ep管中,按照体积比gaba标准溶液:储备溶液a:储备溶液c=10:1:1分别加入储备溶液a和储备溶液c,混合均匀,于40℃水浴条件下反应2h,反应完毕,氮吹干燥并用乙腈水溶液(乙腈与水的体积比为6:4,乙腈水溶液中含有体积分数为0.1%的甲酸)复溶;
lc-ms检测:取gaba衍生化产物进行高效液相色谱-质谱分析。
高效液相色谱的检测条件为:色谱柱:agilentinfinitylabporoshell120ec-c18,规格:2.1×50mm、2.7μm;流动相a为0.1%甲酸(v/v),流动相b为100%乙腈(v/v)(含0.1%甲酸(v/v))。上样量5μl,通过泵以0.3ml/min的流速上样16min,洗脱梯度:0~10min,5~100%b;10~13min,100%b;13~13.1min,100~5%b;13.1~16min,5%b;
质谱的检测条件为:离子源:电喷雾电离源正离子模式;毛细管电压:3500v;干燥气温度:220℃;干燥气流速:8l/min;雾化气气压:199.9kpa。实验仪器使用agilent1260uhplc-6540qtofms联用分析系统。
得到gaba衍生化产物的提取离子色谱图,如图1所示,保留时间从小于1min迁移到4-5min,物质的疏水性明显的到改善;质谱信号响应从小于103增加到105,质谱检测响应提高1-2个数量级。
数据处理:从质谱结果中提取衍生化产物[m+py]+(离子峰m/z=394.1801)的质谱图(如图2所示),从图中可以看出产物峰具有较好的同位素分布模式,且质量偏差处于20ppm以内,也证明了衍生化产物离子的真实存在;
尽管吡喃盐过量尚未有合适条件分离,但是在液相色谱分析中保留时间较大并不干扰gaba衍生物信号(如图3所示),吡喃盐与衍生化产物色谱分离结果互不干扰不影响质谱的离子化效率;
以gaba浓度为横坐标x,衍生化产物所对应的峰面积为纵坐标y,以外标法定量,得到gaba的衍生化产物的标准曲线,如图4所示,在gaba浓度0.5~1000nm/l范围内,标准曲线方程为:y=4684.4613x+277.9387,r=0.99991;
3)小鼠脑提取物中gaba含量的测定
衍生化反应:取100μl小鼠脑提取液于1ml的ep管中,加入10μl储备溶液a(小鼠脑提取液与2,4,5-三苯基吡喃鎓四氟硼酸盐的摩尔比为1:100)、10μl储备溶液c(三乙胺在体系中的浓度为8.33mmol/l),混合均匀,于40℃水浴条件下反应2h,反应完毕,冷却;
lc-ms检测:取反应产物进行高效液相色谱-质谱分析;高效液相色谱和质谱的检测条件同上;
数据处理:从质谱结果中提取对应的衍生化产物的质谱图,通过检测产物所对应的峰面积,由标准曲线得到检测样品中gaba的含量,再计算得到小鼠脑提取物中gaba的含量为19.4nmol/l。
实施例2
基于10种已有吡喃盐标记五种混合单胺类神经递质——多巴胺(da)、组胺(histamine)、5-羟色胺(5-ht)、去甲肾上腺素(ne)和4-氨基丁酸(gaba),并利用lc-ms实现高灵敏质谱检测的方法,包括以下步骤:
1)衍生化试剂、三乙胺溶液及神经递质混合标准溶液的配制
分别准确称取十种吡喃盐(如表1所示)于干燥的试剂小瓶中,加入10ml无水乙腈配制成浓度1mmol/l的储备溶液a,密封后置于冰箱中储存备用;
表1
其中p3、p5、p7、p8、p10为α-活性吡喃盐。
分别准确配制配置5μmmol/l的五种单胺类神经递质(多巴胺、组胺、5-羟色胺、去甲肾上腺素和gaba)溶液,等体积混合得到的混合溶液(每种神经递质的浓度均为1μmmol/l),记为储备溶液b,密封后置于冰箱中储存备用;
准确配制浓度为100mmol/l的三乙胺的乙腈溶液,记为储备溶液c,密封后置于冰箱中储存备用;
取10个干燥的离心管,分别量取储备溶液c配制成浓度为1μmol/l、0.5μmol/l、0.1μmol/l、50nmol/l、20nmol/l、10nmol/l、5nmol/l、1nmol/l、0.8nmol/l、0.5nmol/l的系列浓度的混合标准溶液;
2)衍生化反应
衍生化反应:分别量取100μl系列浓度的混合标准溶液于1.5ml的ep管中,按体积比混合标准溶液:储备溶液a:储备溶液b=10:1:1加入储备溶液a和储备溶液b,混合均匀后于40℃水浴条件下反应2h,反应完毕,冷却;
高效液相色谱的检测条件为:色谱柱:agilentinfinitylabporoshell120ec-c18,规格:2.1×50mm、2.7μm;流动相a为0.1%甲酸(v/v),流动相b为100%乙腈(v/v)(含0.1%甲酸(v/v))。上样量5μl,通过泵以0.3ml/min的流速上样16min,洗脱梯度:0~10min,5~100%b;10~13min,100%b;13~13.1min,100~5%b;13.1~16min,5%b;质谱的检测条件为:离子源:电喷雾电离源正离子模式;毛细管电压:3500v;干燥气温度:220℃;干燥气流速:8l/min;雾化气气压:199.9kpa。实验仪器使用agilent1260uhplc-6540qtofms联用分析系统。
数据处理:从质谱结果中提取混合衍生化产物[m+py]+离子峰(m/z=444.1958,467.2117,460.1907,394.1801,402.1964)的提取离子色谱图,如图5所示,衍生化标记后,五种神经递质保留时间处于3~5min,即使在较低浓度下依然具有较高的信噪比以及明显的信号响应。同时统计了不同吡喃盐通过对衍生物疏水性改变而引起的保留时间偏移(如表2所示),并在图6中对不同吡喃盐的增敏能力进行了统计;
表2
注:nts表示神经递质,即,p1~p10表示衍生物
根据吡喃盐结构的不同,标记后对神经递质理化性质的改变也有明显差异,对于esi离子源检测,疏水性越高(保留时间越大)也就意味着更容易得到更强的质谱信号响应。在十种吡喃盐中,带有较多苯基取代基的吡喃盐(p5-p8)不仅保留时间有更大的改变,对应到图6的质谱信号也有更强的响应,p9由于位阻较大影响了反应产率,因此,为了得到尽可能低的检测限,p5是衍生化标记中的最优选择。
实施例3
几种氨基酸(从accustandardinc购买)衍生化前后采用超高效液相串联四极杆飞行时间质谱uplc-q-tof-ms检测灵敏度对比,具体方法为:
(1)首先将所有氨基酸标准品溶于去离子水中,使最终样品中每种氨基酸浓度均为1μg/ml,超声5min使氨基酸充分溶解。
(2)按照摩尔比为1:50加入适当的吡喃盐,随后将三乙胺乙酸缓冲液(ph=9.5,三乙胺30mm)添加到混合物中并摇动以产生均质的溶液,调节水和乙腈的量,以产生60%乙腈溶液(按体积计)。
(3)最后,混合物在水浴中于50℃加热2小时。反应完成后,将反应混合物在室温下氮气干燥。残留物溶于50%乙腈溶液(v/v)(含0.1%甲酸(v/v))进一步ms分析。
(4)对上述得到的衍生化产物分别进行液相色谱分析,液相色谱条件为:
高效液相色谱的检测条件为:色谱柱:agilentinfinitylabporoshell120ec-c18,规格:2.1×50mm、2.7μm;流动相a为0.1%甲酸(v/v),流动相b为100%乙腈(v/v)(含0.1%甲酸(v/v))。上样量5μl,通过泵以0.3ml/min的流速上样16min,洗脱梯度:0~10min,5~100%b;10~13min,100%b;13~13.1min,100~5%b;13.1~16min,5%b;
质谱的检测条件为:离子源:电喷雾电离源正离子模式;毛细管电压:3500v;干燥气温度:220℃;干燥气流速:8l/min;雾化气气压:199.9kpa。
实验仪器使用agilent1260uhplc-6540qtofms联用分析系统。
表3
注:倍数变化代表衍生化后信号强度与衍生化前信号强度的比值
根据表3结果可以发现,吡喃盐p5和p8能明显增强氨基酸的质谱检测信号,最高增敏幅度可以达到100倍以上,说明吡喃盐可以选择性增敏各种氨基酸,其中碱性氨基酸如精氨酸和组氨酸增敏能力相对较差,苏氨酸增敏最明显,有潜力应用于氨基酸的定性定量分析。
实施例4
几种模板多肽衍生化前后采用超高效液相串联四极杆飞行时间质谱uplc-q-tof-ms检测灵敏度对比,具体方法为:
(1)首先将所有多肽标准品溶于去离子水中,使最终样品中每种多肽浓度均为10μg/ml,超声5min使多肽充分溶解。
(2)按照摩尔比为1:100加入适当的吡喃盐,随后将三乙胺乙酸缓冲液(ph=10.8,三乙胺50mm)添加到混合物中并摇动以产生均质的溶液,调节水和乙腈的量,以产生60%乙腈溶液(按体积计)。
(3)最后,混合物在水浴中于50℃加热3小时。反应完成后,将反应混合物在室温下氮气干燥。残留物溶于50%乙腈溶液(v/v)(含0.1%甲酸(v/v))进一步ms分析。
(4)对上述得到的衍生化产物分别进行液相色谱分析,液相色谱条件为:
高效液相色谱的检测条件为:色谱柱:agilentinfinitylabporoshell120ec-c18,规格:2.1×50mm、2.7μm;流动相a为0.1%甲酸(v/v),流动相b为100%乙腈(v/v)(含0.1%甲酸(v/v))。上样量5μl,通过loading泵以0.3ml/min的流速上样16min,洗脱梯度:0~10min,5~100%b;10~13min,100%b;13~13.1min,100~5%b;13.1~16min,5%b;
质谱的检测条件为:离子源:电喷雾电离源正离子模式;毛细管电压:3500v;干燥气温度:220℃;干燥气流速:8l/min;雾化气气压:199.9kpa。实验仪器使用agilent1260uhplc-6540qtofms联用分析系统。
选用表4中五种多肽用于标记实验(多肽的序列分别为ftyryi;teaek;adlak;tqhfk;tpsek,由安徽省国平药业有限公司合成),分别采用2,4,5-三苯基吡喃盐(p5)、2-苯基-4,5-二对甲基苯基吡喃盐(p7)、2,3,4,5-四苯基吡喃盐(p8)衍生化多肽,标记类型如表4所示。图7给出了2,4,5-三苯基吡喃盐(p5)衍生化多肽的主要方式,所有n端氨基均能被标记,对于含有k的多肽倾向于形成二标记产物[m+2py]2+。
表4五种多肽衍生化后离子类型
通过uplc-q-tof-ms检测,发现了对应m/z,通过积分峰面积得到了衍生化前后多肽的信号强度,为了方便比较,使用相对强度(倍数变化)分析,如图8所示,可以看出,经过衍生化多肽信号强度明显增强最多可以达到一个数量级,表明衍生化对多肽检测具有明显提升能力。
实施例5
对衍生化后多肽的串联质谱结果进行分析
多肽hgtvvl(肌红蛋白水解多肽)参照实施例4中的方法得到相应的衍生化产物。
对上述得到的衍生化产物采用超高效液相串联四极杆飞行时间质谱uplc-q-tof-ms分别进行液相色谱-质谱分析,液相色谱条件为:
高效液相色谱的检测条件为:色谱柱:agilentinfinitylabporoshell120ec-c18,规格:2.1×50mm、2.7μm;流动相a为0.1%甲酸(v/v),流动相b为100%乙腈(v/v)(含0.1%甲酸(v/v))。上样量5μl,通过loading泵以0.3ml/min的流速上样16min,洗脱梯度:0~10min,5~100%b;10~13min,100%b;13~13.1min,100~5%b;13.1~16min,5%b;
质谱的检测条件为:离子源:电喷雾电离源正离子模式;毛细管电压:3500v;干燥气温度:220℃;干燥气流速:8l/min;雾化气气压:199.9kpa;采集模式为automs/ms;质量范围m/z50~3000;碎裂能量20ev。
吡喃盐标记后多肽n端携带有永久正电荷,通常多肽cid解离生成b和y离子,而由于正电荷存在,本申请中多肽cid解离更倾向于生成a和b离子;如图9所示,多肽hgtvvl的ms/ms(二级质谱)图谱中以吡啶正离子m/z308.1559为丰度最强子离子,且a离子和b离子成对出现,m/z差值大致为28,很容易被识别和鉴定,简化了多肽msms图谱的分析。
实施例6
对蛋白水解产物的衍生化结果进行分析
(1)分别使用50mm碳酸氢胺制备1mg/ml的细胞色素c,肌红蛋白和胰蛋白酶溶液(美国sigma)
(2)在37℃下使用胰蛋白酶酶解24h(按照摩尔比,酶:蛋白质=1:50)。之后,将溶液在氮气流下于40℃干燥。干燥后的残留物随后溶解在体积百分数50%的乙腈水溶液中,并在-20℃下保存。
(3)上述溶解液室温解冻后取10μl于ep管,继续添加10mm2,4,5-三苯基吡喃盐(p5)10μl(10mm)震荡均匀后加入20μl三乙胺乙酸缓冲液(ph=10.8,三乙胺50mm),将混合物在50℃的水浴中加热3小时。反应完成后,将样品干燥,然后用于测试。
(4)对上述得到的衍生化产物分别进行液相色谱分析,液相色谱条件为:
高效液相色谱的检测条件为:色谱柱:agilentinfinitylabporoshell120ec-c18,规格:2.1×50mm、2.7μm;流动相a为0.1%甲酸(v/v),流动相b为100%乙腈(v/v)(含0.1%甲酸(v/v))。上样量5μl,通过泵以0.3ml/min的流速上样16min,洗脱梯度:0~10min,5~100%b;10~13min,100%b;13~13.1min,100~5%b;13.1~16min,5%b;
质谱的检测条件为:离子源:电喷雾电离源正离子模式;毛细管电压:3500v;干燥气温度:220℃;干燥气流速:8l/min;雾化气气压:199.9kpa。实验仪器使用agilent1260uhplc-6540qtofms联用分析系统。
图10分别给出了肌红蛋白和细胞色素c酶解产物衍生化前后色谱结果,其中图10a和图10c为肌红蛋白和细胞色素c酶解产物衍生化前的色谱结果,图10b和图10d为肌红蛋白和细胞色素c酶解产物衍生化后的色谱结果,结果表明,衍生化标记后色谱保留时间明显增大(疏水性增强),除此之外信号强度也明显提升,表明衍生化方法对蛋白质的标记同样适用。
选择其中一条细胞色素c水解产生的多肽用于ms/ms分析,cid模式能量为20ev,结果如图11所示,说明α-活性吡喃盐能标记所有多肽上氨基,双标记产物cid裂解更容易形成m/z308.1559,可用于mrm检测,单标记产物能生成规律的ab离子简化序列解析,与采用2,4,6-三苯基吡喃盐(p6)(非α-活性吡喃盐)标记对比能取得更好的解析效果(图11a)。
以上所述,仅是本申请的几个实施例,并非对本申请做任何形式的限制,虽然本申请以较佳实施例揭示如上,然而并非用以限制本申请,任何熟悉本专业的技术人员,在不脱离本申请技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
- 还没有人留言评论。精彩留言会获得点赞!