一种多级萃取法制备脂肪乳样品的前处理方法与流程
本发明属于药物的样品前处理技术领域,具体涉及一种多级萃取法制备脂肪乳样品的前处理方法。
背景技术:
目前,多级萃取在化工领域应用非常广泛,精馏塔是一种典型的多级萃取,通过多级萃取提纯粗品,从而获得高纯度的化工产品;所用溶剂为单一溶剂,在精馏塔的每个塔板上不断反复进行液-液萃取的过程。
由于脂肪乳中含有大量的油脂,既有亲水部分又有亲油部分,给有机物的检测造成极大的困难。目前未见关于不同溶剂的多级萃取的样品前处理方法方面的报道。
技术实现要素:
本发明的目的是提供一种使用多级萃取的前处理方法制备脂肪乳样品以检测脂肪乳中八甲基环四硅氧烷含量的方法。在该方法提供的样品前处理和gc条件下,空白溶液(环己烷)和供试品溶液中八甲基环四硅氧烷无干扰。该检测方法同时还具有准确度、精确度高、重复性好的特点。
本发明提供了如下的技术方案:
一种多级萃取法制备脂肪乳样品的前处理方法,s1、取八甲基环四硅氧烷对照品,用环己烷溶解稀释至0.6μg/ml,作为对照品溶液;
s2、取待测脂肪乳样品,取2ml供试品置50ml比色管中,加200μl乙腈,加入8ml纯水,加入9.8ml乙腈,加入3g氯化钠,剧烈振荡3min;
将溶液全部转移至20ml玻璃顶空瓶中,1000r/min转速离心1.5min,精密量取4ml离心后的上清液置20ml玻璃顶空瓶中,加入4ml纯水,加入4ml环己烷剧烈振荡3min,1000r/min转速离心1.5min,取上清液作为供试品溶液,进样;
s3、平行配制2份对照品溶液,经gc法检测,分别获得对照品溶液中八甲基环四硅氧烷的峰面积,计算八甲基环四硅氧烷峰面积与对照品溶液的浓度的比值,即响应因子。
s4、取供试品溶液,经gc法检测,获得供试品溶液中八甲基环四硅氧烷的峰面积,用响应因子计算供试品溶液中八甲基环四硅氧烷的含量。
优选的,gc法的参数为:色谱柱为agilentdb-1(30m×0.32mm×0.25μm),进样口温度为260℃,载气为n2,流速2ml/min,不分流模式,检测器为fid,检测器温度为260℃,进样体积1μl,升温程序为:50℃保持5min,以10℃/min升至115℃,保持5min,以7℃/min升至150℃,以20℃/min升至230℃,保持1min。
本发明的有益效果:
(1)本发明采用的样品前处理和gc方法测定脂肪乳中八甲基环四硅氧烷的含量,具有如下显著的特点。第一,采用乙腈和环己烷多级萃取脂肪乳,利用极性差异较大的两种溶剂处理脂肪乳,有效地去除了脂肪乳中不同长度碳链的油脂,避免了油脂在气相色谱仪的进样口、色谱柱、检测器等各重要部件的残留,提高了色谱柱的寿命,减少了气相色谱各部件清洗次数和各种耗材的更换,从而大大地节约了成本,有效提高了被测物的准确度。第二,可根据待测物质的检测限度和化合物特性,选择不同的检测器,例如ecd检测器、np检测器和ms检测器等。
(2)在本发明提供的gc条件下,通过外标一点法测定脂肪乳中的八甲基环四硅氧烷含量,在实验室即可实现多级萃取,并可根据化合物特性选择不同的溶剂,同时进行了系列的方法学验证,试验结果表明本发明的检测方法专属性强、准确度好、精密度高、重复性好,符合药物的质量标准研究的技术要求。
附图说明
附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。在附图中:
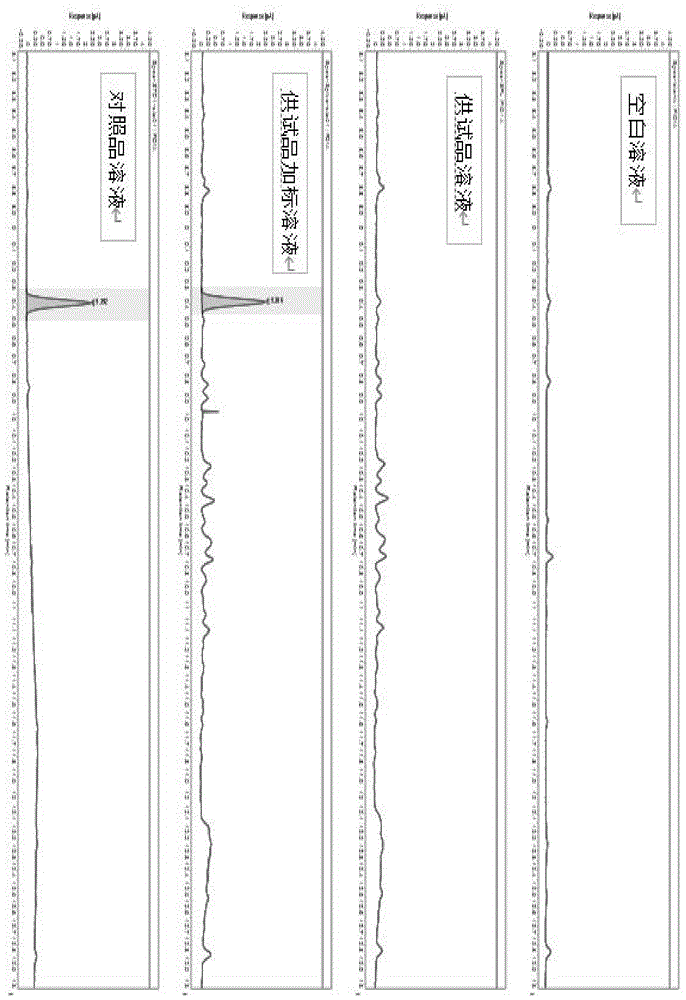
图1为空白溶液、对照品溶液、供试品溶液和供试品加标溶液采集色谱数据;
图2为八甲基环四硅氧烷标准曲线。
具体实施方式
实施例1:
取环己烷作为空白溶液。
精密移取八甲基环四硅氧烷适量,用环己烷溶剂稀释制成每1ml中含有0.6μg八甲基环四硅氧烷的溶液,作为对照品溶液。
精密量取脂肪乳适量2ml,置50ml比色管中,加200μl乙腈,加入8ml纯水,加入9.8ml乙腈,加入3g氯化钠,剧烈振荡3min,将溶液全部转移至20ml玻璃顶空瓶中,1000r/min转速离心1.5min,精密量取4ml离心后的上清液置20ml玻璃顶空瓶中,加入4ml纯水,加入4ml环己烷剧烈振荡3min,1000r/min转速离心1.5min,取上清液作为供试品溶液。
精密量取2ml供试品置50ml比色管中,加入200μl的浓度为30μg/ml的八甲基环四硅氧烷-乙腈溶液,加入8ml纯水,加入9.8ml乙腈,加入3g氯化钠,剧烈振荡3min,将溶液全部转移至20ml玻璃顶空瓶中,1000r/min转速离心1.5min,精密量取4ml离心后的上清液置20ml玻璃顶空瓶中,加入4ml纯水,加入4ml环己烷剧烈振荡3min,1000r/min转速离心1.5min,取上清液作为供试品加标溶液。
gc条件为:色谱柱为agilentdb-1(30m×0.32mm×0.25μm),进样口温度为260℃,载气为n2,流速2ml/min,不分流模式,检测器为fid,检测器温度为260℃,进样体积1μl,升温程序为:50℃保持5min,以10℃/min升至115℃,保持5min,以7℃/min升至150℃,以20℃/min升至230℃,保持1min。
采用本发明提供的gc条件,分别对空白溶液、对照品溶液、供试品溶液和供试品加标溶液采集色谱数据。所得专属性结果如图1所示。
试验结果表明,空白溶液及供试品溶液无干扰,供试品加标溶液与供试品溶液相比,有明显响应。
实施例2:
本实施例提供方法的线性范围考察。
对照品储备溶液1-1(1mg/ml):精密称取50.06mg八甲基环四硅氧烷对照品置50ml容量瓶中,用环己烷稀释至刻度,摇匀。
对照品储备溶液1-2(1mg/ml):精密称取50.09mg八甲基环四硅氧烷对照品置50ml容量瓶中,用环己烷稀释至刻度,摇匀
对照品储备溶液2-1(60μg/ml):精密量取0.6ml对照品储备溶液1-1置10ml量瓶中,用环己烷稀释至刻度,混匀。
对照品储备溶液2-2(60μg/ml):精密量取0.6ml对照品储备溶液1-2置10ml量瓶中,用环己烷稀释至刻度,混匀。
l-1:精密量取1.0ml对照品储备溶液2-1置10ml容量瓶中,用环己烷稀释至刻度,摇匀。
l-2:精密量取0.5ml对照品储备溶液2-1置10ml容量瓶中,用环己烷稀释至刻度,摇匀。
l-3:精密量取0.5ml对照品储备溶液2-1置50ml量瓶中,用稀释剂稀释至刻度,摇匀。
l-4:精密量取5mll-3溶液至10ml容量瓶中,用环己烷稀释至刻度,摇匀。
l-5:精密量取5mll-3溶液至25ml容量瓶中,用环己烷稀释至刻度,摇匀。
将环己烷和各线性点的溶液,分别注入gc中,记录峰面积。试验结果见表1和图2。
表1八甲基环四硅氧烷标准曲线结果
实施例3:
本实施例提供方法的加样回收试验:
通过加样回收的方法,对本发明提供的方法的准确度进行分析。
精密量取脂肪乳适量2ml,置50ml比色管中,加200μl乙腈,加入8ml纯水,加入9.8ml乙腈,加入3g氯化钠,剧烈振荡3min,将溶液全部转移至20ml玻璃顶空瓶中,1000r/min转速离心1.5min,精密量取4ml离心后的上清液置20ml玻璃顶空瓶中,加入4ml纯水,加入4ml环己烷剧烈振荡3min,1000r/min转速离心1.5min,取上清液作为供试品溶液。平行2份。
精密量取2ml供试品置50ml比色管中,加入200μl的浓度为30μg/ml的八甲基环四硅氧烷-乙腈溶液,加入8ml纯水,加入9.8ml乙腈,加入3g氯化钠,剧烈振荡3min,将溶液全部转移至20ml玻璃顶空瓶中,1000r/min转速离心1.5min,精密量取4ml离心后的上清液置20ml玻璃顶空瓶中,加入4ml纯水,加入4ml环己烷剧烈振荡3min,1000r/min转速离心1.5min,取上清液作为供试品加标溶液。平行6份。
按本发明的gc法测定,记录色谱图,按照外标法以峰面积计算回收率,结果见表2~3。结果显示:八甲基环四硅氧烷回收率均在95~105%之间,且回收率rsd为2%。表明本发明提供的方法具有良好的准确度。
表2供试品溶液中八甲基环四硅氧烷含量结果
表3供试品加标溶液结果
实施例4:
本实施例提供方法的稳定性试验
取实施例1中的对照品溶液和供试品加标溶液,于室温下,考察29h内溶液稳定性。按本发明的gc法,分别进样,记录八甲基环四硅氧烷的峰面积,分别计算不同考察点的对照品溶液和供试品加标溶液相对于其0h点的回收率。结果见表4~5。
结果表明29h内,对照品溶液和供试品加标溶液的回收率均在95~105%,说明本发明提供的方法具有良好的稳定性。
表4对照品溶液稳定性结果
表5供试品加标溶液稳定性结果
以上仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!