一种从膜分离过程中污染膜上提取花色苷类物质的方法
本发明涉及花色苷的提取和鉴定技术领域,尤其涉及一种从膜分离过程中污染膜上提取花色苷类物质的方法。
背景技术:
蓝莓和蓝莓提取物因其高含量的花色苷而闻名,花色苷具有很高的抗氧化能力,可以通过中和不稳定的自由基,在预防癌症、心血管和阿尔茨海默症等许多退行性疾病中发挥重要作用。蓝莓是季节性水果,不容易储存,常被加工成果酱、果汁或高含量花色苷的蓝莓提取物。而浓缩蓝莓汁或其提取物是最常见的保存方法,以减少体积或重量,减少包装,便于处理和运输,以及使浓缩液具有较长的保质期。
膜分离技术是用于从生物质加工的产物、副产物和含水或醇的废物加工中回收、分馏和浓缩酚类化合物的有用技术,且已成功用于乳制品、果汁、葡萄酒、植物油、饮用水和农业废水等食品和饮料行业。膜分离技术利用了膜的选择性,一般以进料液和滤液的浓度梯度或操作压力作为推动力,在分子水平上使不同粒径分子的混合物在通过一定截留分子量半透膜时,将大于孔径的大分子物质截留,而使小分子物质穿过膜孔随着溶剂进入滤液中,从而实现选择性分离。膜分离技术作为一种非热分离技术,以其能耗低、工艺步骤少、分离效率高、产品质量好、不同截留分子量的膜可以满足果汁加工过程的澄清、浓缩等不同的分离要求等优点取代了传统的浓缩方法。
膜污染是制约膜分离技术的主要问题。如蓝莓中含有大量酚类物质,特别是花色苷物质,容易与果汁或提取物中存在的其他成分如蛋白质、碳水化合物、脂质等次生代谢物之间发生相互作用,其吸附在膜上后易导致膜通量和分离特性等发生不可逆变化。为了表征和识别污垢,必须首先提取膜上/膜内的化合物。然而,目前尚未有关于从膜材料中提取花色苷类物质的报道,而从植物中提取花色苷类物质的方法不适用于膜材料,原因在于:花色苷与膜结合紧密,且有机污染在膜上易形成复合物,这会增加从污染膜上提取和鉴定花色苷类物质的难度。例如,申请号为cn200610013546.6的中国专利文献公开了一种从黑豆皮中提取花色苷的方法,包括如下步骤:(1)黑豆皮原料的提取:向黑豆皮原料中加入乙醇水溶液或甲醇水溶液,浸泡提取,过滤,所述乙醇水溶液或甲醇水溶液的ph为1-4,浸提液浓缩至无醇,静置,弃沉淀,得上清液;(2)苯乙烯类树脂吸附分离。上述酸性乙醇水溶液或甲醇水溶液对植物中的花色苷具有较好的提取效果,但当用于从膜中提取花色苷时,效果并不理想。
技术实现要素:
为了解决上述技术问题,本发明提供了一种从膜分离过程中污染膜上提取花色苷类物质的方法。通过该方法能从污染膜上提取到花色苷类物质,且提取效果较好。
本发明的具体技术方案为:
一种从膜分离过程中污染膜上提取花色苷类物质的方法,包括以下步骤:
(1)污染膜前处理:将污染膜在25±2℃下避光干燥后,剪切成膜碎片;
(2)提取溶剂制备:将乙腈、甲醇、异丙醇、水、乙酸充分混合后,制备成提取溶剂;
(3)花色苷提取:将膜碎片和提取溶剂在避光容器中混合后,在黑暗中25±2℃下搅拌1-2h,然后离心,收集上清液,即获得花色苷提取液。
在污染膜中,花色苷与膜紧密结合,且易与单糖、多糖、酚酸等物质形成复合物,故相较于从植物中提取花色苷类物质而言,从膜中提取的条件更加苛刻。发明人通过大量实验后,创造性地将乙腈、甲醇、异丙醇、水、乙酸复配作为提取溶剂,能成功地从污染膜中提取花色苷类物质,且提取效果较好;并且,上述提取溶剂中的各成分均对膜中花色苷的提取至关重要,缺少任意一种成分,均会造成提取效果不理想。
此外,花色苷类物质稳定性差,在光和热作用下易降解成酚酸和醛类,因此,本发明的污染膜前处理和花色苷提取过程均在避光和接近室温的条件下进行,从而防止因花色苷降解而导致其鉴定结果不准确。
步骤(1)中,所述污染膜可为超滤膜、纳滤膜、反渗透膜、微滤膜,其材质可为聚醚砜(pes)、聚丙烯腈(pan)、聚偏氟乙烯(pvdf)、聚苯乙烯(ps)、聚酰胺(pa)。
作为优选,步骤(2)中,所述乙腈、甲醇、异丙醇、水、乙酸的体积比为25:25:25:25:0.1-0.5。
进一步地,步骤(2)中,所述乙腈、甲醇、异丙醇、水、乙酸的体积比为25:25:25:25:0.1。
除了提取溶剂的成分外,各成分的配比也会影响污染膜中花色苷的提取效果。发明人通过实验发现,当乙腈、甲醇、异丙醇、水、乙酸的体积比控制在25:25:25:25:0.1-0.5时,从膜中提取到的单体花色苷和聚合花色苷量较大,当体积比控制在25:25:25:25:0.1时,提取效果最好。
作为优选,步骤(3)中,所述膜碎片与提取溶剂的质量体积比为4-10g:100ml。
作为优选,步骤(4)中,获得花色苷提取液后,通过hplc-ms(高效液相色谱-质谱联用技术-质谱联用技术)对花色苷提取液中的花色苷进行鉴定。
作为优选,步骤(1)中,所述干燥方法为自然干燥,干燥时间为18-24h。
作为优选,步骤(3)中,所述搅拌采用磁力搅拌仪,转速为750-1000rpm。
作为优选,步骤(3)中,所述避光容器为棕色瓶子。
作为优选,步骤(3)中,所述离心的转速为5000-6500rpm,温度为4℃,时间10-20min。
作为优选,hplc(高效液相色谱)的操作条件如下:使用反相c-18柱,流动相a为1-5wt%的甲酸水溶液,流动相b为乙腈,流速为1-1.5ml/min,检测波长为520nm。
作为优选,ms(质谱)的操作条件如下:采用esi离子源,正离子灵敏度模式检测;毛细管电压为2.8-3.2kv,样品锥孔电压为25-35v,样品萃取电压为4.8-5.3v,离子源温度为115-125℃,脱溶剂气体温度为345-355℃;喷雾气为高纯氮气,碰撞气体为高纯氩气,反吹气体流速为75-85l/h,脱溶剂气体流速750-850l/h;质谱扫描范围为100-1200da,扫描次数为0.2-0.4s。
与现有技术相比,本发明具有以下优点:
(1)污染膜前处理和花色苷提取过程均在避光和接近室温的条件下进行,能防止因花色苷降解而导致其鉴定结果不准确;
(2)采用乙腈、甲醇、异丙醇、水、乙酸的混合液作为提取溶剂,并将各成分的配比控制在一定范围内,对污染膜中花色苷类物质的提取效果较好。
附图说明
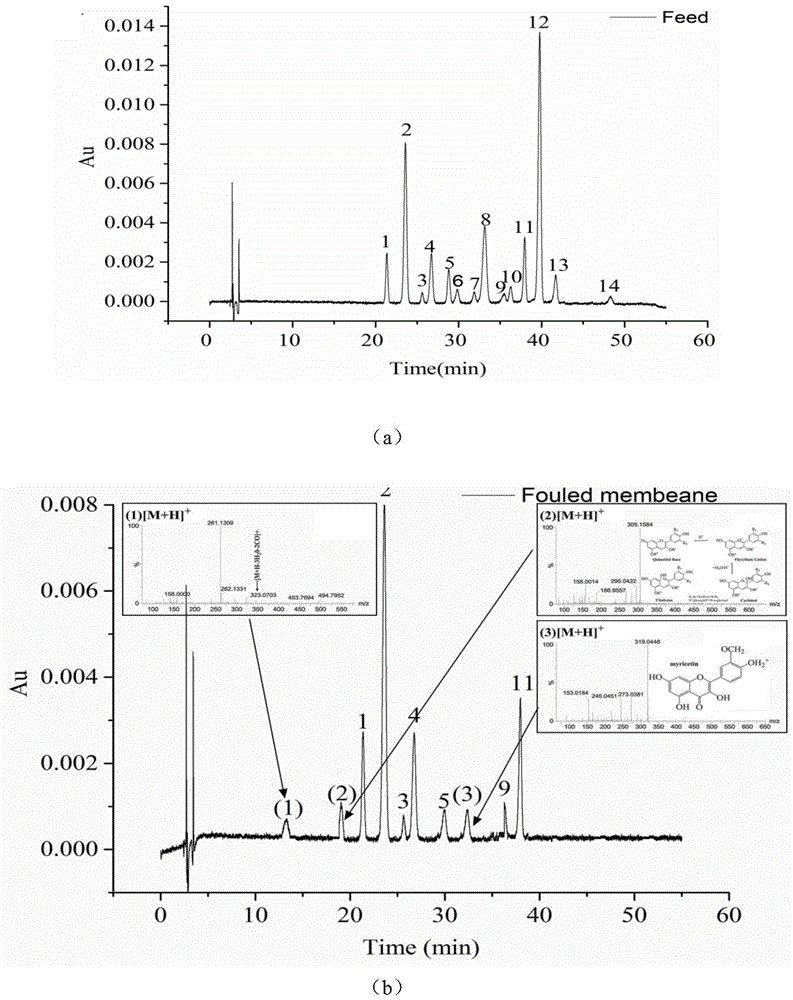
图1为实施例1中花色苷鉴定的质谱图;其中,图(a)为进料液的质谱图,图(b)为花色苷提取液的质谱图;
图2为进料液中14种花色苷单体以及查尔酮、杨梅素的质谱图。
具体实施方式
下面结合实施例对本发明作进一步的描述。
总实施例
一种从膜分离过程中污染膜上提取花色苷类物质的方法,包括以下步骤:
(1)污染膜前处理:将污染膜在25±2℃下避光干燥18-24h后,剪切成膜碎片;
(2)提取溶剂制备:将乙腈、甲醇、异丙醇、水、乙酸按25:25:25:25:0.1-0.5的体积比充分混合后,制备成提取溶剂;
(3)花色苷提取:将膜碎片和提取溶剂按4-10g:100ml的质量体积比在避光容器中混合后,置于采用磁力搅拌仪上,以750-1000rpm的转速在黑暗中25±2℃下搅拌1-2h,然后在2-6℃下以5000-6500rpm的转速离心10-20min,收集上清液,即获得花色苷提取液;
(4)花色苷鉴定:通过hplc-ms对花色苷提取液中的花色苷进行鉴定;
hplc的操作条件如下:使用反相c-18柱,流动相a为1-5wt%的甲酸水溶液,流动相b为乙腈,流速为1-1.5ml/min,检测波长为520nm;
ms的操作条件如下:采用esi离子源,正离子灵敏度模式检测;毛细管电压为2.8-3.2kv,样品锥孔电压为25-35v,样品萃取电压为4.8-5.3v,离子源温度为115-125℃,脱溶剂气体温度为345-355℃;喷雾气为高纯氮气,碰撞气体为高纯氩气,反吹气体流速为75-85l/h,脱溶剂气体流速750-850l/h;质谱扫描范围为100-1200da,扫描次数为0.2-0.4s。
实施例1
污染膜来源于蓝莓提取液的膜浓缩过程,所述膜浓缩过程包括以下步骤:
(i)25g蓝莓干经食物料理机打碎,分散在含有2l纯水的棕色玻璃瓶中,在磁力搅拌器上以750rpm的速率在25℃在黑暗中搅拌提取1.5h,抽滤(采用gb-191490mm直径的滤纸),得到蓝莓水提物粗提液,将粗提液置于冷冻离心机中以5000rpm的速率,离心10min,离心温度为4℃,得到上清液;
(ii)利用pa材质的纳滤膜(400da)在跨膜压力为1.0mpa,流速为1.0l/min,温度25±2℃避光条件将步骤(i)中得到的上清液进行浓缩,得到了体积浓缩系数为2.0的蓝莓提取物浓缩液。
将步骤(ii)中经过纳滤后的污染膜取出后,从污染膜上提取花色苷类物质,包括以下步骤:
(1)污染膜前处理:将污染膜避光自然干燥(温度为25±2℃)24h后,剪切成大小约为3mm×3mm的膜碎片;
(2)提取溶剂制备:将乙腈、甲醇、异丙醇、水、乙酸按25:25:25:25:0.1的体积比充分混合后,制备成提取溶剂;
(3)花色苷提取:将膜碎片和提取溶剂按8g:100ml的质量体积比在棕色瓶子中混合后,置于采用磁力搅拌仪上,以750rpm的转速在黑暗中25℃下搅拌1.5h,然后在4℃下以5000rpm的转速离心10min,收集上清液,即获得花色苷提取液。
通过hplc-ms对进料液(即步骤(ii)中获得的蓝莓提取物浓缩液)和步骤(3)中获得的花色苷提取液中的花色苷进行鉴定,hplc-ms的操作条件如下:
hplc的操作条件:采用waters2695高效液相色谱仪和waters2489紫外检测器;使用zorbaxc18色谱柱(250mm×4.6mm×5μm,agilent,usa),流动相a为5wt%的甲酸水溶液,流动相b为乙腈,流速为1ml/min,梯度方案如下:5%b(0~5min)、5%b(5~15min)、10%b(15~28min)、10%b(28~35min)、13%b(35~50min)、5%b(50~55min);柱恒温25℃,检测波长为520nm,进样量为10mol/l;
ms的操作条件:采用watersuplc-synaptg2联用仪器、esi离子源,正离子灵敏度模式检测;毛细管电压为3.0kv,样品锥孔电压为30v,样品萃取电压为5.0v,离子源温度为120℃,脱溶剂气体温度为350℃;喷雾气为高纯氮气,碰撞气体为高纯氩气,反吹气体流速为80l/h,脱溶剂气体流速800l/h;质谱扫描范围为100-1200da,扫描次数为0.3s;校正曲线标准物质为甲酸钠;实时校正标准物质m/z=556.2771+(亮氨酸脑啡肽);
获得的进料液和花色苷提取液的hplc图分别如图1(a)和图1(b)所示,图1(a)中各峰的质谱图如图2所示。从图2中获得各峰的保留时间和质荷比,并据此确定各峰所代表的单体花色苷种类,结果见表1。
表1花色苷单体的鉴定
对照表1和图1(b),可确定从污染膜上提取到了7种与蓝莓提取物浓缩液中相同的花色苷,分别为飞燕草苷-3-o-半乳糖苷,飞燕草苷-3-o-葡萄糖苷,飞燕草苷-3-o-阿拉伯糖苷,矢车菊素-3-o-半乳糖苷,牵牛花素-3-o-半乳糖苷,芍药花素-3-o-葡糖苷和锦葵色素-3-o-阿拉伯糖苷,另外还检测到查尔酮及杨梅素。
实施例2
从与实施例1中相同的污染膜上提取花色苷类物质,包括以下步骤:
(1)污染膜前处理:将污染膜避光自然干燥(温度为25±2℃)24h后,剪切成大小约为3mm×3mm的膜碎片;
(2)提取溶剂制备:将乙腈、甲醇、异丙醇、水、乙酸按25:25:25:25:0.1的体积比充分混合后,制备成提取溶剂;
(3)花色苷提取:将膜碎片和提取溶剂按4g:100ml的质量体积比在棕色瓶子中混合后,置于采用磁力搅拌仪上,以750rpm的转速在黑暗中25℃下搅拌1.5h,然后在4℃下以5000rpm的转速离心10min,收集上清液,即获得花色苷提取液。
实施例3
本实施例与实施例2的区别在于,步骤(2)中,将乙腈、甲醇、异丙醇、水、乙酸按25:25:25:25:0.3的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
实施例4
本实施例与实施例2的区别在于,步骤(2)中,将乙腈、甲醇、异丙醇、水、乙酸按25:25:25:25:0.5的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
实施例5
本实施例与实施例2的区别在于,步骤(2)中,将乙腈、甲醇、异丙醇、水、乙酸按25:25:25:25:1的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
实施例6
本实施例与实施例2的区别在于,步骤(2)中,将乙腈、甲醇、异丙醇、水、乙酸按30:30:30:10:0.1的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
实施例7
本实施例与实施例2的区别在于,步骤(2)中,将乙腈、甲醇、异丙醇、水、乙酸按20:40:20:20:0.1的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
对比例1
本对比例与实施例2的区别在于,步骤(2)中,将乙醇、水、盐酸混合,制备成提取溶剂,其中,乙醇与水的体积比为20:80,hcl的质量分数为0.1wt%,其他步骤与实施例2相同。
对比例2
本对比例与实施例2的区别在于,步骤(2)中,将甲醇、水、盐酸混合,制备成提取溶剂,其中,甲醇与水的体积比为80:20,hcl的质量分数为0.1wt%,其他步骤与实施例2相同。
对比例3
本对比例与实施例2的区别在于,步骤(2)中,将乙腈、甲醇、异丙醇、水按25:25:25:25的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
对比例4
本对比例与实施例2的区别在于,步骤(2)中,将甲醇、异丙醇、水、乙酸按25:25:25:0.1的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
对比例5
本对比例与实施例2的区别在于,步骤(2)中,将乙腈、异丙醇、水、乙酸按25:25:25:0.1的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
对比例6
本对比例与实施例2的区别在于,步骤(2)中,将乙腈、甲醇、水、乙酸按25:25:25:0.1的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
对比例7
本对比例与实施例2的区别在于,步骤(2)中,将乙腈、甲醇、异丙醇、乙酸按25:25:25:0.1的体积比充分混合后,制备成提取溶剂,其他步骤与实施例2相同。
测试例
对实施例2-7和对比例1-7获得的花色苷提取液中的单体花色苷和聚合花色苷进行测定,获得污染膜中的总单体花色苷和聚合花色苷含量,具体方法如下:
(1)单体花色苷的测定:
将花色苷提取液用氯化钾缓冲液(ph1.0)和醋酸钠缓冲液(ph4.5)稀释。在黑暗中放置30min,用分光光度计分别在520nm和700nm处测定吸光度。tac1的计算公式如下:
式中,mw,花色苷的分子量(矢车菊素-3-葡萄糖苷,449.2g/mol);df,稀释系数;ε,消光系数(26900l/mol·cm);l,1cm。
污染膜中总单体花色苷含量tac2的计算公式如下:
式中,10,萃取膜污垢的溶剂体积,ml;0.4,污染膜质量,g;df,测定tac1时上清液的稀释因子。
(2)聚合花色苷的测定:
将花色苷提取液用纯水稀释,使其在520nm处的吸光度介于0.5~1.0之间。向2.8ml稀释的样品中加入0.9m的焦亚硫酸钾0.2ml,向2.8ml稀释的样品(未漂白的对照样品)中加入0.2ml蒸馏水。静置15min后,样品分别在700nm、520nm和420nm处进行测定。对照样品按下式计算色密度:
colordensity=[(a420nm-a700nm)+(a520nm-a700nm)]×dilutionfactor
用亚硫酸氢盐漂白样品以测定聚合花色苷含量,公式如下:
polymericcolor=[(a420nm-a700nm)+(a520nm-a700nm)]×dilutionfactor
聚合色百分率按下式计算:
%polymericcolor=(polymeirccolor/colordensity)×100
污染膜中总聚合花色苷含量的计算公式如下:
污染膜中的总单体花色苷和聚合花色苷含量测定结果如表2所示。
表2混合溶剂提取膜污垢中花色苷
比较表2中各实施例和对比例的数据,可以得出以下结论:
(1)在从植物中提取花色苷的现有技术中,通常采用酸性的甲醇水溶液或乙醇水溶液作为提取溶剂,对比例1和对比例2中采用这两种提取溶剂对污染膜中的花色苷进行提取,发现获得的单体花色苷和聚合花色苷量均明显低于实施例2,表明这些现有技术中的提取溶剂不适用于从污染膜中提取花色苷类物质。
(2)对比例3-7中采用的提取溶剂中分别少加了一种成分,其提取到的单体花色苷和聚合花色苷量均明显低于实施例2,表明提取溶剂中的各成分均对膜中花色苷的提取至关重要,缺少任意一种成分,均会造成提取效果不理想。
(3)实施例2-7中均采用乙腈、甲醇、异丙醇、水、乙酸的混合溶液作为提取溶剂,各成分之间的配比不同。相较于实施例5-7而言,实施例2-4提取到的单体花色苷和聚合花色苷量较高,其中,实施例2最高,表明当乙腈、甲醇、异丙醇、水、乙酸的体积比控制在25:25:25:25:0.1-0.5时,对污染膜中的花色苷具有较好的提取效果,体积比为25:25:25:25:0.1时的提取效果最好。
本发明中所用原料、设备,若无特别说明,均为本领域的常用原料、设备;本发明中所用方法,若无特别说明,均为本领域的常规方法。
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效变换,均仍属于本发明技术方案的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!