一种盐酸溴己新片溶出度的检测方法与流程
1.本发明涉及一种检测方法,具体涉及一种盐酸溴己新片溶出度的检测方法。
背景技术:
2.盐酸溴己新,英文名称:bromhexinehydrochloride,化学名称:n
‑
甲基
‑
n环己基
‑2‑
氨基
‑
3,5
‑
二溴苯甲胺盐酸盐,结构式为:
[0003][0004]
盐酸溴己新临床应用广泛,用于治疗慢性支气管炎、哮喘等痰粘不易咳出的患者,肠胃吸收快而完全,口服0.5
‑
3小时达到峰值。
[0005]
其中,药物的吸收是口服药物发挥作用关键的第一步,而药物的溶出是吸收的前提条件,因此对盐酸溴己新片进行溶出度检测是非常必要的。
[0006]
目前,针对盐酸溴己新片溶出度的检测方法主要有以下几种:
[0007]
1)chp2020/chp2015盐酸溴己新片溶出度方法:
[0008]
取本品,照溶出度与释放度测定法(中国药典2020年版通则0931第二法),以水900ml为溶出介质,转速为每分钟75转,依法操作,经45分钟时,取溶液10ml,用有机膜滤过,至少弃去初滤液5ml,取续滤液作为供试品溶液;另取盐酸溴己新对照品约16mg,精密称定,置100ml量瓶中,加乙醇4ml振摇使溶解,用水稀释至刻度,摇匀,精密量取1ml,置20ml量瓶中,用水稀释至刻度,摇匀,作为对照品溶液。精密量取供试品溶液和对照品溶液各50μl,照含量测定项下的方法测定,按外标法以峰面积计算每片的溶出量。限度为标示量的70%,应符合规定。
[0009]
其色谱条件与系统适用性:以十八烷基硅烷键合硅胶为填充剂,磷酸盐缓冲液
‑
乙腈(20:80,用0.5mol/l氢氧化钠溶液调节ph值至7.0)为流动相,检测波长为245nm,柱温为40℃,理论板数按盐酸溴己新峰计算应不低于2000,拖尾因子应不大于2.0。
[0010]
2)日本橙皮书盐酸溴己新片溶出度方法:
[0011]
取本品,照溶出度测定法(桨板法),以水900ml为溶剂,转速为每分钟50转,依法操作,经30分钟时,取溶液适量,弃去至少10ml初滤液,精密量取续滤液适量,加流动相稀释1倍,摇匀,作为供试品溶液。另精密称取经105℃干燥4小时的对照品适量,加流动相溶解并稀释制成每1ml中含4μg的溶液,精密量取适量,加水稀释1倍,摇匀,作为对照品溶液。精密量取上述两种溶液各100μl,注入液相色谱仪,记录色谱图;按外标法以峰面积计算溶出量,限度为标示量的75%,应符合规定。
[0012]
其色谱条件与系统适用性:以十八烷基硅烷键合硅胶为填充剂,以0.09%庚烷磺酸钠溶液
‑
甲醇
‑
正丙醇(55:35:10,用10%磷酸调ph值至3.0)为流动相,检测波长为246nm,柱温为40℃,调整流速使盐酸溴己新峰保留时间约为6分钟,理论板数按盐酸溴己新峰计算
应不低于2000,拖尾因子应不大于2.0。
[0013]
然而,经研究发现,上述两种方法中,以水为溶出介质时,滤膜的吸附影响较大,不同来源的水检测结果差异较大,不能真实反映制剂质量,因此,为了更好的体现盐酸溴己新片的溶出吸收情况,研究一种准确性高和可重复性好的盐酸溴己新片溶出度检测方法是十分必要的。
技术实现要素:
[0014]
本发明提供的一种盐酸溴己新片溶出度的检测方法,旨在解决上述背景技术中存在的内容。
[0015]
为了实现上述技术目的,本发明主要采用以下技术方案:
[0016]
一种盐酸溴己新片溶出度的检测方法,包括以下步骤:
[0017]
(1)对照品溶液的制备:取盐酸溴己新对照品适量,用甲醇稀释制成每1ml中含0.4mg的溶液,精密量取适量,用溶出介质为0.05~0.15mol/l的盐酸溶液稀释制成每1ml中含8μg的溶液;
[0018]
(2)供试品溶液的制备:量取经脱气处理的溶出介质0.05~0.15mol/l盐酸溶液,倒入溶出杯内,待温度恒定在37℃
±
0.5℃后,取盐酸溴己新片供试品,投入溶出杯内,控制转速为50rpm,搅拌30分钟后,吸取溶出液10ml,立即用微孔滤膜滤过,取续滤液作为供试品溶液;
[0019]
(3)高效液相色谱法测定:取供试品溶液和对照品溶液各100μl,按照高效液相色谱法进行测定,计算盐酸溴己新片的溶出度。
[0020]
其中,本发明中,在步骤(2)中,所述溶出杯中盐酸溶液的加入量为900ml,所述盐酸溶液浓度为0.1mol/l。
[0021]
本发明中,在步骤(2)中,所述微孔滤膜为有机微孔滤膜,所述有机微孔滤膜的孔径尺寸为0.45μm。
[0022]
本发明中,在步骤(3)中,所述高效液相色谱法的测定条件为,以十八烷基键合硅胶为填充剂,磷酸盐缓冲液
‑
乙腈为流动相,两者体积比为55
‑
65:45
‑
35,进行等度洗脱,流速1.0
‑
1.5ml/min,检测波长245nm,柱温40℃。
[0023]
进一步的,所述磷酸盐缓冲液浓度为0.02mol/l磷酸二氢钾,所述0.02mol/l磷酸二氢钾的ph值为3.2。
[0024]
进一步的,所述磷酸盐缓冲液与乙腈的体积比为60:40。
[0025]
本发明中,上述盐酸溴己新片溶出度的计算公式为
[0026][0027]
其中,
[0028]
a
样
:供试品溶液的溴己新峰峰面积;
[0029]
a
对
:对照品溶液的溴己新峰峰面积;
[0030]
w
对
:盐酸溴己新对照品称取量(mg);
[0031]
p
对
:盐酸溴己新对照品的含量(%);
[0032]
v
样
:溶出介质体积(ml);
[0033]
v
对
:盐酸溴己新对照品稀释倍数;
[0034]
标示量:8mg。
[0035]
进一步的,盐酸溴己新片的溶出度不得低于80%。
[0036]
与现有技术相比,本发明具有以下有益效果:本发明通过优化盐酸溴己新片溶出度检测过程中的条件参数,提高了检测结果的准确性和可重复性。实验结果表明,采用本发明提供的方法对盐酸溴己新片的溶出度进行检测,溶出量提高到80%以上,且rsd较小,更能真实反映制剂质量,同时本发明提供的方法采用0.1mol/l盐酸溶液为溶出介质,与盐酸溴己新片体内释放行为更接近,滤膜无吸附影响,专属性强、重现性好、准确性、稳定性和耐用性均较高,能更好的体现盐酸溴己新片的溶出吸收情况。
具体实施方式
[0037]
以下通过实施例形式对本发明的上述内容再作进一步的详细说明,但不应将此理解为本发明上述主题的范围仅限于以下的实例,凡基于本发明上述内容所实现的技术均属于本发明的范围。
[0038]
实施例1
[0039]
(1)对照品溶液的制备:取盐酸溴己新对照品适量,用甲醇稀释制成每1ml中含0.4mg的溶液,精密量取适量,用0.05~0.15mol/l的盐酸溶液,优选为0.1mol/l的盐酸溶液稀释制成每1ml中含8μg的溶液;
[0040]
(2)供试品溶液的制备:量取经脱气处理的0.05~0.15mol/l盐酸溶液,优选为0.1mol/l的盐酸溶液900ml倒入溶出杯内,待温度恒定在37℃
±
0.5℃后,取盐酸溴己新片供试品6片(或12片),分别投入6个(或12个)溶出杯内,控制转速为50rpm,搅拌30分钟后,吸取溶出液10ml,立即用0.45μm有机微孔滤膜滤过,取续滤液作为供试品溶液;
[0041]
(3)高效液相色谱法测定:取供试品溶液和对照品溶液各100μl,按照高效液相色谱法进行测定,以十八烷基键合硅胶为填充剂,磷酸盐缓冲液
‑
乙腈为流动相,两者体积比为55
‑
65:45
‑
35,优选两者体积比为60:40,进行等度洗脱,流速1.0
‑
1.5ml/min,优选流速为1.0ml/min,检测波长245nm,柱温40℃,根据溶出度计算公式,计算盐酸溴己新片的溶出度。溶出度的计算公式如下:
[0042][0043]
其中,
[0044]
a
样
:供试品溶液的溴己新峰峰面积;
[0045]
a
对
:对照品溶液的溴己新峰峰面积;
[0046]
w
对
:盐酸溴己新对照品称取量(mg);
[0047]
p
对
:盐酸溴己新对照品的含量(%);
[0048]
v
样
:溶出介质体积(ml);
[0049]
v
对
:盐酸溴己新对照品稀释倍数;
[0050]
标示量:8mg。
[0051]
对比例1
[0052]
按照本发明实施例1的方法进行溶出度计算,其不同之处在于溶出介质分别为
ph4.5醋酸盐缓冲液。
[0053]
对比例2
[0054]
按照本发明实施例1的方法进行溶出度计算,其不同之处在于溶出介质分别为ph5.5磷酸盐缓冲液。
[0055]
对比例3
[0056]
按照本发明实施例1的方法进行溶出度计算,其不同之处在于溶出介质分别为ph6.8磷酸盐缓冲液。
[0057]
(1)比较盐酸溴己新在实施例1与对比例1
‑
3中不同介质中的稳定性
[0058]
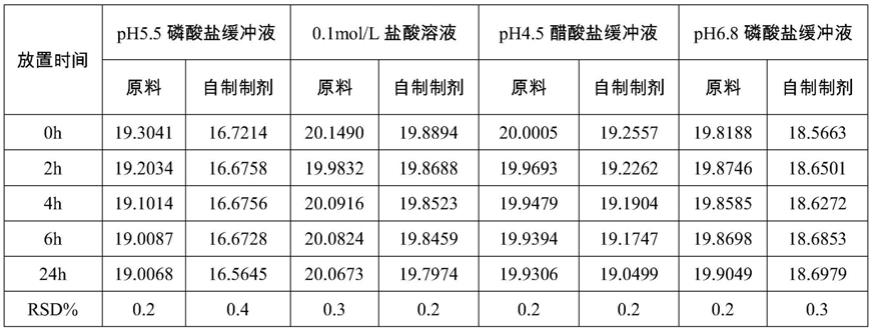
取盐酸溴己新原料和自制的盐酸溴己新片剂细粉适量,用溶出介质(0.1mol/l盐酸溶液、ph4.5醋酸盐缓冲液、ph6.8磷酸盐缓冲液和ph5.5磷酸盐缓冲液)溶解并稀释制成溶液,室温下放置,每隔一段时间进样测定,计算样品峰面积的rsd,结果见下表1:
[0059]
表1:盐酸溴己新在4种介质中的稳定性试验结果
[0060][0061]
结论:盐酸溴己新原料和自制的盐酸溴己新片在0.1mol/l盐酸溶液、ph4.5醋酸盐缓冲液、ph6.8磷酸盐缓冲液和ph5.5磷酸盐缓冲液溶出介质中24小时内稳定。
[0062]
(2)溶出介质的选择
[0063]
申请人采用中试放大批自制制剂(批号:20190503)对比了不同来源的水介质中的溶出度(参照chp2020/2015盐酸溴己新片的溶出条件),结果如下表2。
[0064]
表2:盐酸溴己新在3种水中的溶出度试验结果
[0065]
来源溶出度%rsd%自制纯化水77.65.9超纯水105.95.4娃哈哈水79.76.2
[0066]
结论:超纯水和纯化水的溶出度结果相差约28%,差异很大。因此水不适合作为溶出介质。
[0067]
另外,本品在自制纯化水中,滤膜吸附较为严重,且不同浓度的盐酸溴己新溶液不呈线性关系,采用自制纯化水作为溶出介质进行方法学验证时发现,线性和回收率均达不到可接受标准。线性结果如下表3。
[0068]
表3:盐酸溴己新采用自制纯化水作为溶出介质的线性结果
[0069][0070][0071]
回收率结果如下表4。
[0072]
表4:盐酸溴己新采用自制纯化水作为溶出介质的回收率结果
[0073][0074]
结论:盐酸溴己新在自制纯化水中,在4.093~9.822μg/ml浓度范围内(相当于测定浓度的51.16%~122.78%),峰面积与浓度不呈线性关系(r=0.9968)。
[0075]
盐酸溴己新在自制纯化水中,50%水平的平均回收率为74.0%,在80%水平的平均回收率为91.7%,均不符合可接受标准。因此不适合选择水作为溶出介质。
[0076]
同时,申请人通过溶出度实验发现,盐酸溴己新片在ph4.5醋酸盐缓冲液、ph5.5磷酸盐缓冲液、ph6.8磷酸盐缓冲液中,均无法达到终溶。因此,选择0.1mol/l盐酸溶液作为标准介质,ph4.5醋酸盐缓冲液、ph5.5磷酸盐缓冲液、ph6.8磷酸盐缓冲液以及纯化水作为辅助介质进行溶出曲线检测。
[0077]
同时对参比制剂(批号:171318)在900ml的5种溶出介质中溶出曲线测定,结果见下表5。
[0078]
表5:参比制剂在不同溶出介质中溶出曲线比较结果
[0079][0080]
备注:0.1mol/l盐酸溶液、ph4.5醋酸盐缓冲液和ph6.8磷酸盐缓冲液中转速均为50rpm。
[0081]
结果分析:在0.1mol/l盐酸溶液中15min大于85%,因此选择0.1mol/l盐酸溶液作为标准介质;在ph4.5醋酸盐缓冲液中,45min大于85%;在ph6.8磷酸盐缓冲液中无法完全溶出,45min达到终溶平台(约30%),在ph5.5磷酸盐缓冲液和纯化水中溶出释放行为均受水来源的影响较大。因此选择0.1mol/l盐酸溶液作为标准介质,选择ph4.5醋酸盐缓冲液、ph5.5磷酸盐缓冲液和ph6.8磷酸盐缓冲液作为辅助介质进行溶出曲线检测。并采用娃哈哈水,转速选择75rpm,在稳定性0天和末期进行溶出曲线对比研究。ph5.5磷酸盐缓冲液和水介质的溶出曲线因受水来源影响较大,均不作为溶出相似性评价依据。
[0082]
(3)色谱条件的选择
[0083]
chp2020/chp2015和日本橙皮书盐酸溴己新片质量标准中溶出量的测定方法中流动相略有差异,因此采用4种介质配制的对照品溶液对色谱条件进行了对比研究,结果如下:
[0084]
a、采用chp2020/chp2015盐酸溴己新片质量标准中溶出量测定方法
[0085]
色谱柱为agilent zorbax sb
‑
c18 4.6
×
50mm,5μm;流动相为磷酸盐缓冲液(取磷酸二氢钾1.0g,用水900ml使溶解,用0.5mol/l氢氧化钠溶液调ph值至7.0,加水至1000ml)
‑
乙腈(20:80),进样4种介质配制的对照品溶液,其中0.1mol/l盐酸溶液配制的对照品溶液,主峰未出峰,且该流动相为ph7.0体系,对色谱柱损伤较大,故未采用该色谱系统进行溶出量的测定。
[0086]
b、采用日本橙皮书盐酸溴己新片溶出量测定方法
[0087]
色谱柱为agilent zorbax sb
‑
c18 4.6
×
50mm,5μm;流动相为0.09%庚烷磺酸钠溶液
‑
甲醇
‑
正丙醇(55:35:10,用10%磷酸调ph值至3.0),进样4种介质配制的对照品溶液,4种介质条件配制的溶液均能出峰,可采用该方法检测溶出量,但该方法采用离子对试剂作为流动相,对色谱柱有一定的损伤,且为了日常检测方便,故未采用该色谱系统进行溶出量的测定。
[0088]
c、采用含量测定的流动相
[0089]
含量测定方法的流动相为0.02mol/l磷酸二氢钾缓冲液(取磷酸二氢钾2.74g,用水1000ml使溶解,加三乙胺1ml,用磷酸调ph值至3.2)
‑
乙腈(60:40),采用该方法,进样4种介质配制的对照品溶液均能出峰,可采用该方法检测溶出量,且该方法与含量测定方法一致,便于后期的质量研究,因此选择该方法作为溶出量检测方法。
[0090]
(4)滤膜吸附考察
[0091]
取样过滤时,可能滤膜存在吸附影响而导致主成分损失,故需进行滤膜吸附的验证。
[0092]
1)用溶出介质分别配制对照品溶液,分别分成7份,1份直接进样,平行测定6次;另6份用滤膜过滤,弃初滤液3ml后,再每次过滤3ml,收集6份续滤液,取续滤液各进样1针,记录色谱图。计算过滤前后峰面积rsd。
[0093]
2)因chp2020/chp2015中溶出液过滤时采用有机膜,故增加有机膜与水系膜的滤膜吸附对比。其中滤膜信息如表6所示,滤膜吸附实验结构如表7、表8、表9、表10所示。
[0094]
表6滤膜信息
[0095]
名称材质规格来源滤膜1mce0.45μm
×
25mmwelch滤膜2疏水ptfe0.45μm
×
25mmwelch滤膜3聚醚砜0.22μm
×
25mm津腾滤膜4尼龙0.22μm
×
25mm津腾
[0096]
表7:滤膜吸附试验结果
‑
对照品溶液(水系膜
‑
welch mce滤膜)
[0097][0098][0099]
结果分析:采用welch的mce水系滤膜,纯化水、ph4.5醋酸盐缓冲液和ph6.8磷酸盐
缓冲液下,过滤前后峰面积rsd均大于2%,说明该材质滤膜对盐酸溴己新主成分有吸附。
[0100]
表8:滤膜吸附试验结果
‑
对照品溶液(有机膜
‑
welch ptfe疏水滤膜)
[0101][0102]
结果分析:采用welch的ptfe有机系滤膜,纯化水和ph6.8磷酸盐缓冲液下,过滤前后峰面积rsd均大于2%,0.1mol/l盐酸溶液、ph4.5醋酸盐缓冲液和ph5.5磷酸盐缓冲液下,弃初滤液3ml,过滤前后峰面积rsd均小于2%。说明该材质滤膜在纯化水和ph6.8磷酸盐缓冲液下,对盐酸溴己新主成分有吸附;在0.1mol/l盐酸溶液、ph4.5醋酸盐缓冲液和ph5.5磷酸盐缓冲液条件下,对盐酸溴己新主成分几乎无吸附。
[0103]
表9:滤膜吸附试验结果
‑
对照品溶液(津腾滤膜)
[0104][0105]
结果分析:采用津腾的水系和有机系滤膜,纯化水和ph6.8磷酸盐缓冲液下,过滤前后峰面积rsd均大于2%,ph5.5磷酸盐缓冲液下,过滤前后峰面积rsd小于2%。说明该材质滤膜在纯化水和ph6.8磷酸盐缓冲液下,对盐酸溴己新主成分有吸附;在ph5.5磷酸盐缓冲液条件下,对盐酸溴己新主成分几乎无吸附。
[0106]
取ph6.8磷酸盐缓冲液的原料配成2.2μg/ml(本品终溶的浓度)溶液,取经浸泡饱和过的滤膜连续过滤,分别采用玻璃注射器和塑料注射器,取过滤前后的溶液各进样1针,记录色谱图,计算连续过滤峰面积rsd。结果如表6所示。
[0107]
表10:滤膜吸附试验结果
‑
对照品溶液(不同注射器)
[0108]
[0109][0110]
结果分析:采用津腾有机系滤膜,用玻璃注射器,弃12ml初滤液后,滤膜对主成分几乎无吸附。
[0111]
通过以上表3
‑
表6的结果,得出以下滤膜吸附结论:
[0112]
0.1mol/l盐酸溶液配制的对照品和供试品溶液采用welch疏水ptfe滤膜,弃滤液3ml后,滤膜对盐酸溴己新主成分几乎无吸附。ph4.5醋酸盐缓冲液和ph5.5磷酸盐缓冲液配制的对照品和供试品溶液采用welch疏水ptfe滤膜,弃滤液3ml后滤膜对盐酸溴己新主成分几乎无吸附。但是为了尽量减少检测误差,实际操作中选择弃初滤液6ml后进行试验。
[0113]
ph5.5磷酸盐缓冲液配制的对照品和供试品溶液,采用津腾尼龙滤膜,弃滤液3ml后,滤膜对盐酸溴己新主成分几乎无吸附,但是为了尽量减少检测误差,实际操作中选择去初滤液6ml后进行试验。
[0114]
ph6.8磷酸盐缓冲液配制的对照品和供试品溶液,采用津腾有机系滤膜,预先经浸泡饱和后晾干,用玻璃注射器,弃12ml初滤液后,滤膜对主成分几乎无吸附。
[0115]
水介质配制的对照品和供试品溶液,采用welch疏水ptfe滤膜,津腾尼龙滤膜和津腾聚醚砜滤膜,均有吸附,其中津腾尼龙滤膜吸附最小。
[0116]
对比实验
[0117]
取三批次(每批次取供试品6片)的盐酸溴己新片,分别采用原中国药典2015版或2020版方法与采用本发明实施例1提供的方法,对盐酸溴己新的溶出度进行检测,并对每批次的检测结果取平均值,其结果如下表1所示。
[0118]
表1溶出度进行结果
[0119][0120]
其中,中国药典2015版或2020版数据引用纯化水介质溶出曲线中45min溶出量数据。
[0121]
结论:变更前后方法检测对比,变更后方法溶出量较变更前方法高,且rsd较小,更能真实反映制剂质量。
[0122]
溶出度检查分析方法学验证,其结果如下表7所示。
[0123]
表7:溶出度检查分析方法学验证结果
[0124]
[0125][0126][0127]
结论:表面本发明提供的测定方法具有专属性强、重现性好、准确性、稳定性和耐用性均较高,能更好的体现盐酸溴己新片的溶出吸收情况。
[0128]
上述实施例仅为本发明的优选实施方式之一,不应当用于限制本发明的保护范围,但凡在本发明的主体设计思想和精神上作出的毫无实质意义的改动或润色,其所解决
的技术问题仍然与本发明一致的,均应当包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1