一种稳心中药组合物的检测方法与流程
本发明涉及一种稳心中药组合物的检测方法,属于药品质量检测方法领域。
背景技术:
1、本发明所述的稳心中药组合物可以为稳心颗粒、稳心片,该产品是由山东步长制药股份有限公司生产,其主要由党参、黄精、三七、琥珀、甘松等成分制备而成。其该药的功效为:益气养阴,活血化瘀。用于气阴两虚,心脉瘀阻所致的心悸不宁,气短乏力,胸闷胸痛;室性早搏、房室早搏见上述证候者。它临床上用于稳定心率失常等疾病,有着显著的临床疗效显著。本技术人对该产品进行了布局多件该产品的相关专利申请,申请号分别为:01131734.5、200410081398.2、200610042976.0、200610042974.1、200810232536.0、200810183930.x、201510145198.7、201610013406.2、201811308826.9、201910483321.4,上述专利保护内容主题大多集中在从组方、制备工艺改进、质量检测成分含量检测、有效物质成分研究、防治变态反应性疾病、骨折临床新用途等。
2、对我公司产品的质量检测方法进行系统梳理可知,cn101088549a公开采用十八烷基硅烷键合相硅胶为填充剂;乙腈-水为流动相,梯度洗脱条件:0-18分钟:乙腈-水配比为1∶9,18-30分钟:乙腈-水配比为34∶66,30-38分钟:乙腈-水配比为9∶1,38-50分钟:乙腈-水配比为1∶9;检测波长为203nm;柱温为30℃;流速为1.0ml/min,并测定三七皂苷r1、人参皂苷rg1、人参皂苷rb1含量。然而cn104730193a公开一种稳心颗粒中挥发性成分的检测方法,采用顶空固相微萃取技术,提取稳心颗粒中挥发性成分。并结合利用气相色谱-质谱联技术,共分离鉴定出挥发性成分74个。cn105548425a公开一种采用高效液相法测定三七皂苷r1、人参皂苷rg1、人参皂苷rb1含量的步骤,该方法还包括了采用高效液相方法测定人参皂苷rd与党参炔苷含量的步骤。本发明在利用高效液相法测定三七皂苷r1、人参皂苷rg1、人参皂苷rb1含量的基础上增加了高效液相法测定人参皂苷rd与党参炔苷的含量,使稳心颗粒的质量控制的指标更加全面;本发明检测方法的精密度、稳定性、重现性好。但上述检测方法在工艺化大生产的过程中,还存在操作步骤复杂、按照现行标准中规定的色谱条件进行试验人参皂苷rg1与干扰峰re未达到有效分离,且供试品制备过程中,弃去的碱液中含有部分目标成分,导致测得含量低于实际值。因此,研发开发一种操作简便、准确度高、且分离度好的检测方法显得尤为必要。
技术实现思路
1、本发明目的在于,提供一种稳心中药组合物的检测方法,该检测方法具有操作简便可行、专属性强、稳定性、重复性可靠,可作为本发明稳心中药组合物的内在质量检测方法。
2、本发明中药组合物检测方法的技术方案具体如下:
3、一种稳心中药组合物的检测方法,所述检测方法包括以下步骤:
4、⑴、供试品溶液的制备:取本发明中药,称定,加入70~90%甲醇,超声处理,再称定重量,用70~90%甲醇补足减失的重量,摇匀,滤过,即得;
5、⑵、对照品溶液的制备:取三七皂苷r1对照品、人参皂苷rg1对照品和人参皂苷rb1对照品适量,精密称定,加甲醇制成混合溶液,即得;
6、⑶、色谱条件为:色谱柱:十八烷基硅烷键合硅胶为填充剂,流动相:a相为乙腈-b相为水,其梯度洗脱程序为:0-12min,流动相a的体积配比16→19%,流动相b的体积配比84→81%;12~35min,流动相a的体积配比19→24%,流动相b的体积配比81→76%;35~38min,流动相a的体积配比24→28%,流动相b的体积配比76→72%;38~60min,流动相a的体积配比28→31%,流动相b的体积配比72→69%;60~70min,流动相a的体积配比31→43%,流动相b的体积配比69→57%;所述检测柱温度:20~30℃,检测波长190~210nm;
7、⑷、测定法:分别吸取对照品溶液和供试品溶液,注入液相色谱仪,测定,即得。
8、优选的,所述检测方法步骤⑴供试品溶液样品的制备中所述甲醇的浓度为80%。
9、优选的,所述检测方法步骤⑴供试品溶液样品的制备中所述超声处理的功率为400~600w、频率为30~50khz、超声时间为30~60min。
10、优选的,所述检测方法步骤⑴供试品溶液样品的制备中所述超声处理的功率为500w、频率为40khz、超声时间为45min。
11、优选的,所述检测方法步骤⑵对照品溶液的制备中所述三七皂苷r1的浓度为0.0878~0.7024μg/ml、人参皂苷rg1的浓度为0.2830~2.2640μg/ml、人参皂苷rb1的浓度为0.2198~1.7584μg/ml。
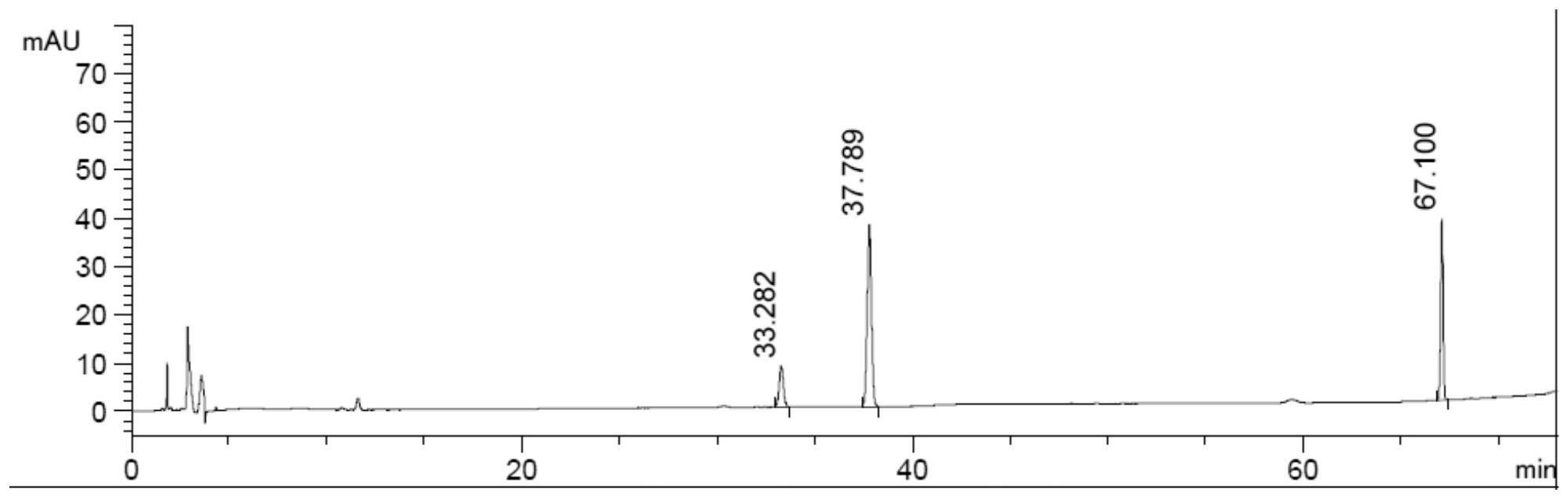
12、优选的,所述检测方法步骤⑶的色谱条件为:所述检测柱温度:25℃,检测波长203nm。
13、优选的,所述检测方法步骤⑶的色谱条件为:所述色谱柱的型号为:un-techtabithathermo bds-c18、喆分zafex supfex jx-c18。
14、为了体现突出本发明技术方案创新性,我们将本实验过程中优选的部分实验提供如下。
15、1色谱条件的确定
16、1.1流动相的选择:
17、《中国药典》2015年版一部三七、稳心片和稳心胶囊项下含量测定的色谱条件均以乙腈-水为流动相进行梯度洗脱,采用稳心片、稳心胶囊项下流动相进行测定,结果发现稳心颗粒中人参皂苷rb1色谱峰与附近干扰峰未达到有效分离,影响定量结果的准确性,故根据本品种的测定情况对流动相配比进行调节,最终确定以乙腈为流动相a,以水为流动相b,按下表中的规定进行梯度洗脱。
18、表1本发明检测方法色谱流动相比例
19、
20、1.2检测波长的选择:
21、原标准的检测方法和《中国药典》2015年版一部稳心片和稳心胶囊项下含量测定的检测波长均为203nm。
22、1.3柱温的选择
23、参照稳心片和稳心胶囊项下,柱温设定为25℃,按照上述流动相进行梯度洗脱,3个成分色谱峰分离效果良好,无干扰;试验发现当柱温设定为30℃、35℃时,人参皂苷rg1与干扰峰分离不佳,故参照稳心片和稳心胶囊方法,规定含量测定的柱温为25℃。
24、1.4耐用性试验
25、使用三种色谱柱:un-tech tabitha5μm,4.6×250mm;thermo bds-c18 5μm,250×4.6mm;喆分zafex supfex jx-c18 5μm,4.6×250mm;按上述色谱条件进行测试;结果表明,三七皂苷r1、人参皂苷rg1和人参皂苷rb1色谱峰的峰形较好,试验条件适用范围广。
26、表2色谱柱的考察结果表(样品批号:2005017)
27、
28、1.5色谱条件及系统适用性试验综上所述,
29、色谱条件为:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,按下表进行梯度洗脱,流速:1ml/min,柱温25℃。理论板数按人参皂苷rg1峰计算应不低于6000。
30、2供试品溶液制备方法的考察
31、2.1对照品溶液的制备:
32、精密称取三七皂苷r1(批号:110745-201318,以94.0%计)19.46mg,置25ml量瓶中,加甲醇溶解并稀释至刻度,摇匀,备用;精密称取人参皂苷rg1(批号:110703-201832,以92.4%计)19.14mg,置10ml量瓶中,加甲醇溶解并稀释至刻度,摇匀,备用;精密称取人参皂苷rb1对照品(批号:110704-201827,以91.2%计)20.08mg,置10ml量瓶中,加甲醇溶解并稀释至刻度,摇匀,备用。
33、分别精密量取上述三七皂苷r1对照品溶液3ml、人参皂苷rg1对照品溶液4ml、人参皂苷rb1对照品溶液3ml,置同一50ml量瓶中,加甲醇稀释至刻度,摇匀,即得对照品混合溶液((三七皂苷r1浓度为0.0439mg/ml、人参皂苷rg1浓度为0.1415mg/ml、人参皂苷rb1浓度为0.1099mg/ml))。
34、2.2提取方法的考察
35、稳心颗粒包含有蔗糖和无蔗糖两种制剂规格,方法学考察所用样品为山东步长制药有限公司提供批号为2005017(平均装量为8.9771g/袋)和2005052(无蔗糖)(平均装量为5.0366g/袋),下同。
36、考察了超声直接提取和碱洗两种提取方式,具体试验情况如下:
37、方法一(原标准方法):取装量差异项下的本品,研细,取约2g或lg(无庶糖),精密称定,置具塞锥形瓶中,精密加入水饱和的正丁醇50ml,密塞,称定重量,浸泡12小时,超声处理(功率300w,频率40khz)1小时,放冷,再称定重量,用水饱和的正丁醇补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用2%氢氧化钠溶液洗涤2次,每次30ml,弃去碱液,再用正丁醇饱和的水30ml洗涤,弃去洗涤液,正丁醇液蒸干,残渣加甲醇溶解并转移至10ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
38、方法二:取装量差异项下的本品,研细,取约2g或lg(无庶糖),精密称定,置具塞锥形瓶中,精密加入水饱和的正丁醇50ml,密塞,称定重量,浸泡12小时,超声处理(功率500w,频率40khz)1小时,放冷,再称定重量,用水饱和的正丁醇补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用浓氨试液洗涤2次,每次30ml,合并碱液,再用水饱和正丁醇溶液洗涤2次,每次25ml,合并正丁醇提取液和洗涤液,蒸干,残渣加甲醇溶解并转移至10ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
39、方法三:取装量差异项下的本品,研细,取约2g或lg(无庶糖),精密称定,置具塞锥形瓶中,分别精密加入甲醇、80%甲醇和60%甲醇25ml,密塞,称定重量,超声处理(功率500w,频率40khz)45分钟,取出,放冷,再称定重量,分别用甲醇、80%甲醇和60%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
40、分别精密吸取2.1项下对照品溶液及上述供试品溶液各10μl,按确定的色谱条件进行测定,结果见表3和表4。
41、表3提取方法考察结果-稳心颗粒2005017
42、
43、表4提取方法考察结果-稳心颗粒2005052(无蔗糖)
44、
45、
46、由测定结果可知,原标准提取方法测得总含量明显低于其他方法,超声提取方法测得总量稍高于碱洗后碱液反洗的方法,采用超声提取直接进样,三七皂苷r1、人参皂苷rg1和人参皂苷rb1均能达到有效分离,故为了减化操作,选择直接超声提取。甲醇、80%甲醇和60%甲醇提取效率无明显差异,由于稳心颗粒2005017样品甲醇超声提取的供试品溶液在24h内有颗粒状物质析出,故提取溶剂选择80%甲醇。
47、2.3提取方式考察
48、超声提取方法;取装量差异项下的本品,研细,取约2g或lg(无庶糖),精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,超声处理(功率500w,频率40khz)45分钟,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
49、回流提取方法:取装量差异项下的本品,研细,取约2g或lg(无庶糖),精密称定,置圆底烧瓶中,精密加入80%甲醇25ml,密塞,称定重量,水浴回流提取1小时,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
50、分别精密吸取2.1项下对照品溶液及上述供试品溶液各10μl,按确定的色谱条件进行测定,结果见表5和表6
51、表5提取方式考察结果-稳心颗粒2005017
52、
53、表6提取方式考察结果-稳心颗粒2005052(无蔗糖)
54、
55、
56、由测定结果可知,超声提取和回流提取没有明显差异,为简化操作,选择超声提取方式。
57、2.4提取时间的考察
58、取装量差异项下的本品,研细,取约2g或lg(无庶糖),精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,分别超声处理(功率500w,频率40khz)20、30、45和60分钟,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
59、分别精密吸取2.1项下对照品溶液及上述供试品溶液各10μl,按确定的色谱条件进行测定,结果见表7和表8。
60、表7提取时间考察结果-稳心颗粒2005017
61、
62、表8提取时间考察结果-稳心颗粒2005052
63、
64、由测定结果可知,为保证提取完全,确定提取方法为超声提取45分钟。
65、2.5取样量的考察
66、取装量差异项下的本品,研细,分别取约1.5g、2g、2.5g或0.5g、lg、1.5g(无庶糖),精密称定,置具塞锥形瓶中,精密入加80%甲醇25ml,密塞,称定重量,超声处理(功率500w,频率40khz)45分钟,取出,放冷,再称定重量,用80%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
67、分别精密吸取2.1项下对照品溶液及上述供试品溶液各10μl,按确定的色谱条件进行测定,结果见表9和表10。
68、表9取样量考察结果-稳心颗粒2005017
69、
70、表10取样量考察结果-稳心颗粒2005052
71、
72、由测定结果可知,为保证提取完全以及取样的均匀性,确定稳心颗粒取样量为2g或1g(无蔗糖)。
73、本发明检测方法的有益效果:
74、1.与原有标准相比较,本发明检测方法中供试品制备步骤大为简化,具体为:将原来“水饱和的正丁醇”提取溶剂更换为“80%甲醇”,以及将原来“浸泡12小时后超声,碱洗后碱液反洗”步骤优化为“超声提取”,这使得原有的操作步骤得以简便,可行,且操作时间大为减少。
75、2.本发明还将原有的色谱条件进行优化,最终得到最佳的色谱条件。其梯度洗脱程序为:0-12min,流动相a的体积配比16→19%,流动相b的体积配比84→81%;12~35min,流动相a的体积配比19→24%,流动相b的体积配比81→76%;35~38min,流动相a的体积配比24→28%,流动相b的体积配比76→72%;38~60min,流动相a的体积配比28→31%,流动相b的体积配比72→69%;60~70min,流动相a的体积配比31→43%,流动相b的体积配比69→57%,并使得人参皂苷rb1色谱峰与附近干扰峰达到有效分离。
76、3.本发明检测方法学考察,精密度试验结果:三七皂苷峰面积r1 rsd(%)为0.96,人参皂苷rg1峰面积rsd(%)为1.21,人参皂苷rb1峰面积rsd(%)为1.09。稳定性试验结果:三七皂苷峰面积r1 rsd(%)为1.76,人参皂苷rg1峰面积rsd(%)为1.23,人参皂苷rb1峰面积rsd(%)为1.53。这表明,供试品溶液在25小时内稳定性良好。重复性试验结果:三七皂苷峰面积r1 rsd(%)为1.57~1.79,人参皂苷rg1峰面积rsd(%)为1.05~1.98,人参皂苷rb1峰面积rsd(%)为0.97~1.32。这表明,该检测方法重复性良好。样品的加样回收率试验结果可知:三七皂苷的r1平均回收率(%)为97.86~98.33,rsd(%)为1.74~1.87。人参皂苷rg1的平均回收率(%)为95.47~95.86,rsd(%)为1.74~1.68,人参皂苷rb1峰面积rsd(%)为95.53~95.69,rsd(%)为1.73~1.99。这表明,该检测方法加样回收率良好。综上所述,本发明检测方法的重新性、稳定性、可靠性良好,可以作为本发明稳心中药组合物内在质量检测方法,并应于该产品的产业化生产质量内控方法。
- 还没有人留言评论。精彩留言会获得点赞!