一种快速净化固相萃取柱及其应用、咖啡制品的前处理方法和丙烯酰胺的检测方法与流程
1.本发明涉及分析检测技术领域,具体涉及一种快速净化固相萃取柱及其应用、咖啡制品的前处理方法和丙烯酰胺的检测方法。
背景技术:
2.丙烯酰胺被国际癌症研究机构(iarc)归类为2a级致癌物,被证明在人体中具有神经毒性,在实验动物中具有生殖毒性和遗传毒性。近年来咖啡越来越受到消费者的喜爱。咖啡属于高温烘焙热加工食品,在其焙烤加工过程产生丙烯酰胺,且含量较高,引起消费者、生产者、管理层关注。2017年11月20日欧盟委员会发布2017/2158号令,制定减少食品中丙烯酰胺含量的缓解措施和基准水平的规范,欧盟对食品中丙烯酰胺含量限定范围大多在300~850μg/kg之间,其中烘焙咖啡为400μg/kg、速溶咖啡为850μg/kg。24个国家获得的2002~2004年间食品中丙烯酰胺的检测数据共6752个,检测的数据包含早餐谷物、土豆制品、咖啡及其制品、奶类等主要消费食品,其中含量较高的三类食品为:高温加工的土豆制品(包括薯片、薯条等),平均含量为0.48mg/kg,最高含量为5.3mg/kg;咖啡及其制品,平均含量为0.51mg/kg,最高含量为7.3mg/kg;早餐谷物类食品,平均含量为0.31mg/kg,最高含量为7.8mg/kg;其它种类食品的丙烯酰胺含量基本在0.1mg/kg以下。
3.目前,咖啡制品中丙烯酰胺检测过程中采用的净化柱主要有hlb柱、max柱、mcx柱、carb柱、al2o3柱、c18柱、bond elut-accucat柱、isolutemultimode柱,isolute env+柱等,然而,上述净化柱一般采用2次或更多柱净化,净化效率低。
4.食品中丙烯酰胺的测定主要是气相色谱质谱法(gc-ms)、液相色谱串联质谱法(lc-ms/ms),如现行的食品中丙烯酰胺的标准检测方法主要有gb5009.204-2014《食品安全国家标准-食品中丙烯酰胺的测定》和sn/t 2096-2008《食品中丙烯酰胺的检测方法-气相色谱质谱同位素内标法》。gb 5009.204-2014中包括两种lc-ms/ms和gc-ms两种检测方法,lc-ms/ms法以水为提取溶剂,经hlb固相萃取柱和bond elut-accucat固相萃取柱或基质分散固相萃取净化,以lc-ms/ms检测,内标法定量,检测限为10μg/kg。但在实际咖啡(尤其是速溶咖啡)样品检测中,由于丙烯酰胺分子量小(分子量为71),极性大,杂质干扰大,检测灵敏度低。欧盟bs iso 18862:2016采用水为提取溶剂,正己烷脱脂,水相经铁氰化钾试剂沉淀后,再经c18固相萃取柱和离子交换固相萃取柱2次净化,稀释后进液相色谱-串联质谱分析;gc-ms法为经溴试剂衍生后,经极性阳离子交换柱进一步净化后进gc-ms分析,然而,lc-ms/ms法检测由于其稀释倍数较大及定性离子72》44灵敏度低,导致方法检测灵敏度低。因此,提供一种灵敏度高的丙烯酰胺的检测方法具有重要的意义。
技术实现要素:
5.本发明的目的在于提供一种快速净化固相萃取柱及其应用、咖啡制品的前处理方法和丙烯酰胺的检测方法,本发明提供的快速净化固相萃取柱能够实现咖啡的快速、简便、
高效净化和降低基质干扰现象,本发明提供的前处理方法处理后样品杂质干扰小,检测准确度和灵敏度高。
6.为了实现上述发明目的,本发明提供以下技术方案:
7.本发明提供了一种快速净化固相萃取柱,其特征在于,包括柱体,所述柱体内设置有两块筛板,所述两块筛板之间装填有填料,所述填料包括混合型阴离子交换填料和混合型阳离子交换填料。
8.优选的,所述填料包括:混合型阴离子交换填料和层叠设置在所述混合型阴离子交换填料表面的混合型阳离子交换填料,或,混合型阳离子交换填料和层叠设置在所述混合型阳离子交换填料表面的混合型阴离子交换填料,或混合型阴离子交换填料和混合型阳离子交换填料的混合物。
9.优选的,所述填料的粒径为10~100μm;
10.所述填料中混合型阴离子交换填料和混合型阳离子交换填料的质量比为(1~4):(1~4)。
11.优选的,所述混合型阳离子交换填料包括divinylbenzene/vinylpyrrolidone+so3h;
12.所述混合型阴离子交换填料包括divinylbenzene/vinylpyrrolidone+ch2n
+
r3,其中,r为烷基。
13.本发明提供了上述技术方案所述的快速净化固相萃取柱在咖啡制品的前处理中的应用。
14.本发明提供了一种咖啡制品的前处理方法,包括以下步骤:
15.将待测咖啡制品、
13
c3d
3-丙烯酰胺内标应用液和水混合,进行提取,得到的提取物进行脱脂,得到样品提取液;
16.采用上述技术方案所述的快速净化固相萃取柱对所述样品提取液进行水淋洗净化,得到样品净化液;
17.将所述样品净化液进行乙腈盐析浓缩,将所得浓缩物复溶于甲酸水溶液中,得到待测样品液。
18.优选的,所述提取为涡旋提取,所述涡旋提取的速度为1500~2500r/min,时间为1~15min。
19.本发明提供了一种咖啡制品中丙烯酰胺的检测方法,包括以下步骤:
20.采用液相色谱串联质谱对上述技术方案所述前处理方法得到的待测样品液中丙烯酰胺进行定性和定量检测。
21.优选的,所述液相色谱串联质谱检测中液相色谱检测的条件包括:
22.色谱柱:联苯基色谱柱;
23.柱温:30℃;
24.进样量:1~20μl;
25.流动相:流动相a和流动相b,所述流动相a为甲酸水溶液,所述甲酸水溶液中甲酸的体积分数为0.1~0.3%;所述流动相b为甲醇,所述流动相的流速为0.2~0.4ml/min;
26.洗脱方式:梯度洗脱;
27.所述梯度洗脱的程序如下:
28.0.00~6.00min,所述流动相a的体积分数为80~95%;
29.6.00~6.10min,所述流动相a的体积分数由80~95%匀速降低到0~5%;
30.6.10~8.00min,所述流动相a的体积分数为0~5%;
31.8.00~8.10min,所述流动相a的体积分数由0~5%匀速增加到80~95%,所述流动相12.00min,所述流动相a的体积分数为80~95%。
32.优选的,所述液相色谱串联质谱检测中质谱检测的条件包括:
33.离子源:电喷雾离子源;
34.毛细管电压:0.5~3.5kv;
35.离子源温度:120~180℃;
36.脱溶剂温度:300~600℃;
37.扫描方式:正离子扫描;
38.检测方式:多反应检测;
39.雾化气:由氮气发生器产生的氮气或高纯氮气;
40.锥孔反吹气:由氮气发生器产生的氮气或高纯氮气;
41.碰撞气:高纯氩气。
42.本发明提供了一种快速净化固相萃取柱,包括柱体,所述柱体内设置有两块筛板,所述两块筛板之间装填有填料,所述填料包括混合型阴离子交换填料和混合型阳离子交换填料。本发明提供的快速净化固相萃取柱只需一次过柱净化,经过简单的水淋洗和水洗脱,即可高效快速去除咖啡制品中的色素、酸性物质、碱性物质等大量杂质,从而降低了在液相色谱串联质谱(lc-ms/ms)检测中的基质干扰现象,提高了后续的lc-ms/ms检测丙烯酰胺的准确度和灵敏度。而且,采用本发明提供的快速净化固相萃取柱进行净化操作快速、简便,大幅提升了工作效率;同时减少了大量有机溶剂的使用,更加绿色、环保。
43.本发明提供了一种咖啡制品的前处理方法,包括以下步骤:将待测咖啡制品、
13
c3d
3-丙烯酰胺内标应用液和水混合,进行提取,得到的提取物进行脱脂,得到样品提取液;采用上述技术方案所述的快速净化固相萃取柱对所述样品提取液进行水淋洗净化,得到样品净化液;将所述样品净化液进行乙腈盐析浓缩,将所得浓缩物复溶于甲酸水溶液中,得到待测样品液。本发明提供的前处理方法,以水为提取剂对于丙烯酰胺的提取率高,安全环保;二氯甲烷脱脂能够将脂肪提取到二氯甲烷层中,丙烯酰胺留在水溶液中,实现脱脂的目的,经快速净化固相萃取柱经水淋洗及洗脱一次净化即可高效快速去除咖啡制品中的色素、酸性物质、碱性物质等大量杂质,从而降低了在lc-ms/ms检测中的基质干扰现象,乙腈盐析浓缩中乙腈的加入能够实现丙烯酰胺的富集,提高检测的灵敏度,经过乙腈盐析浓缩能够进一步大幅降低了在液质检测中的基质干扰现象,提高了后续的lc-ms/ms检测丙烯酰胺的准确度和灵敏度。本发明提供的前处理方法更加简单方便,可操作性强,得到的分析溶液澄清透明,基质干扰低,适合各类咖啡制品中丙烯酰胺的检测。
44.本发明提供了一种咖啡制品中丙烯酰胺的检测方法,包括以下步骤:采用液相色谱串联质谱对上述技术方案所述前处理方法得到的待测样品液中丙烯酰胺进行定性和定量检测。与
13c3-丙烯酰胺为内标物质相比,本发明以以
13
c3d
3-丙烯酰胺做内标物质,定量检测的准确度更高,检测结果更可靠;本发明提供的检测方法简便、快速、检出限低、准确度高。如实施例结果所示,当样品质量为2.0g时,丙烯酰胺的检出限为3μg/kg,定量限为10μg/
kg。
45.进一步的,本发明采用的联苯基色谱柱的保留性能更强,可有效分离干扰基质,进一步提高了lc-ms/ms检测丙烯酰胺的准确度和灵敏度。
附图说明
46.图1为三种装填方式得到的快速净化固相萃取柱的结构示意图;
47.图2为丙烯酰胺及其内标(
13
c3d
3-丙烯酰胺)的标准离子图谱;
48.图3为丙烯酰胺的标准曲线;
49.图4为fapas速溶咖啡质控样品离子谱图;
50.图5为速溶黑咖啡粉样品离子谱图;
51.图6为实施例6中速溶黑咖啡粉样品离子谱图;
52.图7为对比例3的速溶黑咖啡粉样品离子谱图。
具体实施方式
53.本发明提供了一种快速净化固相萃取柱,包括柱体,所述柱体内设置有两块筛板,所述两块筛板之间装填有填料,所述填料包括混合型阴离子交换填料和混合型阳离子交换填料。在本发明中,所述筛板的厚度优选为0.5~5mm,所述筛板的直径与柱体内径匹配;所述的筛板孔的孔径优选小于填料粒径,具体优选为10~100μm。
54.在本发明中,若没有特殊说明,所采用的试剂均为本领域技术人员所熟知的市售商品。
55.在本发明中,所述填料优选包括:混合型阴离子交换填料和层叠设置在所述混合型阴离子交换填料表面的混合型阳离子交换填料,或,混合型阳离子交换填料和层叠设置在所述混合型阳离子交换填料表面的混合型阴离子交换填料,或混合型阴离子交换填料和混合型阳离子交换填料的混合物。在本发明中,所述填料的粒径优选为10~100μm。在本发明中,所述填料中混合型阴离子交换填料和混合型阳离子交换填料的质量比优选为(1~4):(1~4),更优选为(1~3):(1~3),进一步优选为(1~2):(1~2)。在本发明中,所述混合型阳离子交换填料优选包括divinylbenzene/vinylpyrrolidone+so3h,更优选为welchrom p-scx、waters mcx或纳普mcx,进一步优选为waters mcx。在本发明中,所述混合型阴离子交换填料优选包括divinylbenzene/vinylpyrrolidone+ch2n
+
r3,其中,r为烷基,更优选为welchrom p-sax、waters max或纳普max,进一步优选为watersmax;所述快速净化固相萃取柱中填料的质量与柱体的体积之比优选为0.5~1.0g:6ml。
56.在本发明中,所述快速净化固相萃取柱的制备方法优选包括以下步骤:在柱体内设置下筛板,将填料装填到所述下筛板表面,在所述填料上方设置上筛板,得到快速净化固相萃取柱。在本发明中,所述填料装填优选包括分层装填或混合装填,所述分层装填优选包括:在下筛板表面装填混合型阴离子交换填料后装填混合型阳离子交换填料,或,在下筛板表面装填混合型阳离子交换填料后装填混合型阴离子交换填料;所述混合装填优选包括将混合型阴离子交换填料和混合型阳离子交换填料混合后装填到下筛板表面;三种装填方式得到的快速净化固相萃取柱的结构示意图如图1所示。
57.本发明提供了上述技术方案所述的快速净化固相萃取柱在咖啡制品的前处理中
的应用。
58.本发明提供了一种咖啡制品的前处理方法,包括以下步骤:
59.将待测咖啡制品、
13
c3d
3-丙烯酰胺内标应用液和水混合,进行提取,得到的提取物进行脱脂,得到样品提取液;
60.采用上述技术方案所述的快速净化固相萃取柱对所述样品提取液进行水淋洗净化,得到样品净化液;
61.将所述样品净化液进行乙腈盐析浓缩,将所得浓缩物复溶于甲酸水溶液中,得到待测样品液。
62.本发明将待测咖啡制品、
13
c3d
3-丙烯酰胺内标应用液和水混合,进行提取,得到的提取物进行脱脂,得到样品提取液。
63.在本发明中,所述待测咖啡制品优选包括咖啡豆或速溶咖啡粉。在本发明中,当所述待测咖啡制品为咖啡豆时,所述待测咖啡制品在使用前优选进行粉碎。本发明对于所述粉碎的方式没有特殊限定,采用本领域技术人员熟知的粉碎方式粉碎至20~100μm即可。
64.在本发明中,所述
13
c3d
3-丙烯酰胺内标应用液的浓度优选为5.0μg/ml;在本发明中,所述
13
c3d
3-丙烯酰胺内标应用液中
13
c3d
3-丙烯酰胺标准品的质量为待测咖啡制品质量的0.1~2ppm,更优选为0.5~1.5ppm,进一步优选为1ppm。
65.本发明对于所述
13
c3d
3-丙烯酰胺内标应用液的配制方法没有特殊限定,采用本领域技术人员熟知的配制方式即可。在本发明的具体实施例中,所述
13c3-丙烯酰胺内标应用液的配制方法优选包括以下步骤:将
13
c3d
3-丙烯酰胺标准品5.0mg(精确至0.01mg)置于10ml棕色容量瓶中,用乙腈溶解后定容至刻度,得到
13
c3d
3-丙烯酰胺内标储备溶液,-20℃下避光密封保存;准确吸取0.10ml
13
c3d
3-丙烯酰胺储备溶液置于10ml棕色容量瓶中,利用0.1%(v/v)甲酸水溶液定容至刻度,得到
13
c3d
3-丙烯酰胺内标应用液,于4℃下保存。
66.在本发明中,所述待测咖啡制品的质量和水总量(
13
c3d
3-丙烯酰胺内标应用液中的水和额外加入的水)的质量比优选为1:(5~20),更优选为1:(10~15)。
67.在本发明中,所述提取的方式优选为涡旋提取;所述提取的温度优选为室温;所述涡旋提取的速度优选为1500~2500r/min,更优选为2000~2500r/min,所述涡旋提取的时间优选为1~15min,更优选为5~10min。
68.在本发明中,所述脱脂采用的脱脂剂优选包括二氯甲烷或正己烷,所述待测咖啡制品的质量和脱脂剂的体积之比优选为1g:(2~20)ml,更优选为1g:(10~15)ml;所述脱脂的温度优选为室温,所述脱脂的时间优选为5~10min,更优选为7~8min。
69.所述脱脂完成后,本发明优选还包括将所述脱脂体系进行涡旋处理和离心分离,所得上清液为样品提取液。在本发明中,所述涡旋处理的温度优选为室温,所述涡旋处理的速度优选为1500~2500r/min,更优选为2000~2500r/min,所述涡旋处理的时间优选为1~15min,更优选为5~10min;所述涡旋处理的目的是提高丙烯酰胺的提取效率。在本发明中,所述离心分离的速度优选为5000~15000r/min,更优选为10000~12000r/min;所述离心分离的时间优选为1~5min,更优选为2~3min。
70.得到样品提取液后,本发明采用上述技术方案所述的快速净化固相萃取柱对所述样品提取液进行水淋洗净化,得到样品净化液。
71.在本发明中,所述快速净化固相萃取柱在使用前优选进行活化处理,所述活化处
理优选包括;依次使用甲醇和水进行淋洗;所述快速净化固相萃取柱中填料、甲醇和水的用量比优选为1g:6~12ml:6~12ml,更优选为1g:6~8ml:8~10ml。
72.在本发明中,所述水淋洗净化优选包括水淋洗除杂和水洗脱,所述样品提取液、水淋洗除杂用水和水洗脱用水的体积比优选为1:(3~4):(5~8),更优选为1:(3.5~4):(5~6)。
73.得到样品净化液后,本发明将所述样品净化液进行乙腈盐析浓缩,将所得浓缩物复溶于甲酸水溶液中,得到待测样品液。
74.在本发明中,所述乙腈盐析浓缩优选包括:将样品净化液、乙腈、氯化钠和无水硫酸镁混合,进行乙腈盐析提取后离心分离,将所得液位组分进行浓缩。在本发明中,所述样品净化液、乙腈、氯化钠和无水硫酸镁的用量比优选1ml:5~10ml:1~2g:4~6g,更优选为1ml:8~10ml:1~1.5g:4~5g;本发明加入乙腈能够萃取出丙烯酰胺从而实现丙烯酰胺的富集,提高检测的灵敏度,针对含量较高的咖啡和速溶咖啡样品,不进行富集也能满足检测要求。在本发明中,所述乙腈盐析提取优选为涡旋提取,所述涡旋提取的温度优选为室温,所述涡旋提取的速度优选为1500~2500r/min,更优选为2000~2500r/min,所述涡旋提取的时间优选为10~30min,更优选为10~20min。在本发明中,所述离心分离的速度优选为3000~10000r/min,更优选为5000~8000r/min;所述离心分离的时间优选为2~5min,更优选为3~4min。在本发明中,所述浓缩的方式优选为氮气吹干,所述氮气吹干优选在水浴中进行,所述水浴的温度优选为20~50℃,更优选为30~40℃。
75.在本发明中,所述浓缩物与甲酸水溶液的用量比优选为1g:0.1~1ml,更优选为1g:0.5~1ml;所述甲酸水溶液中甲酸的体积分数优选为0.1~0.3%,更优选为0.1~0.2%;所述甲酸水溶液的作用是将丙烯酰胺及其内标物质溶解出来。
76.本发明提供了一种咖啡制品中丙烯酰胺的检测方法,包括以下步骤:
77.采用液相色谱串联质谱对上述技术方案所述前处理方法得到的待测样品液中丙烯酰胺进行定性和定量检测。
78.在本发明中,所述液相色谱串联质谱检测优选包括以下步骤:
79.将所述待测样品液进行液相色谱串联质谱检测,得到样品色谱图;
80.根据所述样品色谱图得到丙烯酰胺和
13
c3d
3-丙烯酰胺的峰面积;根据所述峰面积与标准曲线,采用内标法计算待测样品液中丙烯酰胺的浓度,所述标准曲线为丙烯酰胺的色谱相对峰面积与丙烯酰胺浓度的标准曲线。
81.本发明将所述待测样品液进行液相色谱串联质谱检测,得到样品色谱图。在本发明中,所述液相色谱串联质谱检测中液相色谱检测的条件优选包括:色谱柱优选为联苯基色谱柱,更优选为chromcore biphenyl色谱柱;柱温优选为30℃;进样量优选为1~20μl,更优选为5~10μl;流动相优选为流动相a和流动相b,所述流动相a优选为甲酸水溶液,所述甲酸水溶液中甲酸的体积分数优选为0.1~0.3%,更优选为0.1~0.2%;所述流动相b优选为甲醇;所述流动相的流速优选为0.2~0.4ml/min,更优选为0.3ml/min;洗脱方式优选为梯度洗脱。在本发明中,所述梯度洗脱过程优选如下:
82.0.00~6.00min,所述流动相a的体积分数优选为80~95%,更优选为85~90%;
83.6.00~6.10min,所述流动相a的体积分数优选由80~95%匀速降低到0~5%,更优选为降低至0~2%;
84.6.10~8.00min,所述流动相a的体积分数优选为0~5%,更优选为0~2%;
85.8.00~8.10min,所述流动相a的体积分数优选由0~5%匀速增加到80~95%,更优选增加到85~90%;
86.所述流动相12.00min,所述流动相a的体积分数优选为80~95%,更优选为85~90%。
87.在本发明中,所述液相色谱串联质谱检测中质谱检测的条件包括:离子源优选为电喷雾离子源(esi+);离子源温度优选为120~180℃,更优选为150℃;毛细管电压优选为0.5~3.5kv,更优选为3.0kv;脱溶剂温度优选为300~600℃,更优选为500℃;扫描方式优选为正离子扫描;检测方式优选为多反应检测(mrm);雾化气优选为由氮气发生器产生的氮气高纯氮气,所述高纯氮气的纯度优选为99.999%;锥孔反吹气优选为由氮气发生器产生的氮气高纯氮气,所述高纯氮气的纯度优选为99.999%;碰撞气优选为高纯氩气,所述高纯氩气的纯度优选为99.999%;使用前优选调节所述雾化气、锥孔反吹气和碰撞气的流量以使质谱灵敏度达到检测要求;锥孔电压、碰撞能量等电压值优选优化至最优灵敏度;定性离子对、定量离子对、锥孔电压及碰撞能量如表1所示。
88.表1丙烯酰胺和
13
c3d
3-丙烯酰胺的质谱参数
[0089][0090]
在本发明中,定性分析过程中,每种待测化合物的质谱定性离子必须出现,至少应包括一个母离子和两个子离子,而且同一检验批次,对同一丙烯酰胺,进样液中丙烯酰胺的两个子离子的相对丰度比与浓度相当的标准溶液相比,其允许偏差不超过表2规定的范围:
[0091]
表2定性时相对离子丰度的最大允许偏差
[0092]
相对离子丰度/%>5020-5010-20≤10允许相对偏差/%
±
20
±
25
±
30
±
50
[0093]
得到样品色谱图后,本发明根据所述样品色谱图得到丙烯酰胺和
13
c3d
3-丙烯酰胺的峰面积比值;根据所述峰面积比值与浓度绘制标准曲线,采用内标法计算待测样品液中丙烯酰胺的浓度;所述标准曲线为丙烯酰胺的色谱相对峰面积与丙烯酰胺浓度的标准曲线。
[0094]
在本发明中,所述标准曲线的制作优选包括以下步骤:
[0095]
配制丙烯酰胺系列标准工作溶液;
[0096]
将所述丙烯酰胺系列标准工作溶液进行lc-ms/ms检测,得到丙烯酰胺峰面积与
13
c3d
3-丙烯酰胺峰面积比值与丙烯酰胺浓度的标准曲线。
[0097]
本发明配制丙烯酰胺系列标准工作溶液。在本发明中,所述丙烯酰胺系列标准工作溶液中丙烯酰胺标准品的浓度优选依次为1.0、2.0、5.0、10、20、50、100和200ng/ml,
13
c3d
3-丙烯酰胺的浓度均优选为100ng/ml。在本发明中,所述丙烯酰胺系列标准工作溶液的配制方法优选包括以下步骤:将丙烯酰胺标准品、
13
c3d
3-丙烯酰胺和甲酸水溶液混合后
定容,得到丙烯酰胺系列标准工作溶液;所述甲酸水溶液中甲酸的体积分数优选为0.1~0.3%,更优选为0.1~0.2%。
[0098]
得到丙烯酰胺系列标准工作溶液后,本发明将所述丙烯酰胺系列标准工作溶液进行lc-ms/ms检测,得到丙烯酰胺峰面积与
13
c3d
3-丙烯酰胺峰面积比值与丙烯酰胺浓度的标准曲线。
[0099]
在本发明中,所述lc-ms/ms的检测条件优选与前文所述待测定样品的液相色谱检测、质谱检测的条件一致,在此不再赘述。
[0100]
在本发明中,所述lc-ms/ms检测优选由低浓度到高浓度的顺序进行检测,以丙烯酰胺标准品的色谱峰面积与
13
c3d
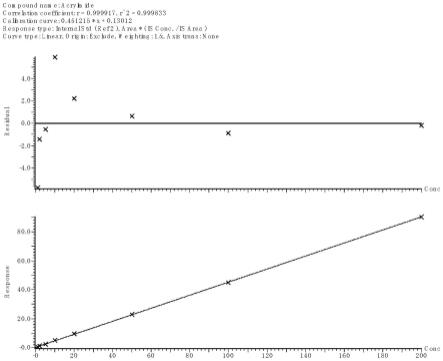
3-丙烯酰胺色谱峰面积之比为纵坐标(y),以丙烯酰胺的浓度为横坐标(x)绘制标准曲线,标准曲线:y=0.451215x+0.13012,r=0.9999。
[0101]
在本发明中,所述标准曲线的线性范围优选为丙烯酰胺浓度1~200ng/ml(含
13
c3d
3-丙烯酰胺浓度为100ng/ml)。
[0102]
在本发明中,样品待测液的响应值在标准曲线线性范围内,当超过线性范围上限时,优选减少取样量或者适当增加内标加入量后重新测定。
[0103]
在本发明中,丙烯酰胺的浓度计算式如式(1)所示:
[0104][0105]
式(1)中,x为样品中丙烯酰胺的含量,单位为μg/kg;
[0106]
c为样品中丙烯酰胺按照内标法在标准曲线中对应的浓度,单位为ng/ml;
[0107]
v为样品待测液的定容体积,单位为ml;
[0108]
f为样品提取液与经快速净化固相萃取柱净化的样品提取液的体积比值;
[0109]
m为样品质量,单位为g。
[0110]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0111]
以下实施例采用的材料与方法如下:
[0112]
样品和试剂:丙烯酰胺(购自德国dr.ehrenstorfer公司);
13
c3d
3-丙烯酰胺标准品(购自美国sigma公司);混合型阴离子交换填料和混合型阳离子交换填料(均购自美国waters公司,waters mcx,waters max,粒径为10~100μm);乙腈、甲醇、二氯甲烷(均购自merck,美国),甲酸为色谱纯(购自美国acs公司),氯化钠为分析纯(购自上海国药公司),无水硫酸镁为分析纯(购自cnw公司),超纯水(milli-q,美国millipore公司);咖啡、速溶咖啡样品均购自杭州超市。
[0113]
仪器:超高效液相色谱串联质谱仪(uplc-ms/ms,美国waters公司);chromcore biphenyl色谱柱(3μm,3.0mm
×
150mm)(纳普科技有限公司);多样品漩涡振荡器(德国heidolph);高速离心机(美国beckman公司);固相萃取仪(cnw公司,快速净化固相萃取柱使用时的一个配套装置),氮吹仪(上海安谱公司),分析天平(感量0.01g,mettler toledo公司);超声波清洗器(昆山超声仪器有限公司)。
[0114]
实施例1
[0115]
(1)标准溶液的配制
[0116]
丙烯酰胺标准储备溶液(1.0mg/ml):准确称取丙烯酰胺标准品10mg(精确至0.01mg),于10ml棕色容量瓶中,乙腈溶解并定容至刻度,-20℃下避光密封保存。
[0117]
丙烯酰胺标准储备溶液(10mg/l):准确吸取1.0mg/ml丙烯酰胺标准储备溶液0.25ml置于25ml棕色容量瓶中,乙腈定容至刻度,-20℃下避光密封保存。
[0118]
丙烯酰胺标准使用液(1.0mg/l):准确吸取10mg/l丙烯酰胺标准储备溶液1.00ml置于10ml棕色容量瓶中,0.1%(v/v)甲酸水溶液定容至刻度,现配现用。
[0119]
13
c3d
3-丙烯酰胺内标储备溶液(0.5mg/ml):准确称取
13
c3d
3-丙烯酰胺标准品5.0mg(精确至0.01mg),于10ml棕色容量瓶中,乙腈溶解并定容至刻度,-20℃下避光密封保存。
[0120]
13
c3d
3-丙烯酰胺内标应用液(5.00mg/l):准确吸取0.10ml
13
c3d
3-丙烯酰胺储备溶液置于10ml棕色容量瓶中,0.1%(v/v)甲酸水溶液定容至刻度,4℃下保存。
[0121]
丙烯酰胺系列标准工作溶液:取适量丙烯酰胺标准使用液(1.0mg/l),以0.1%(v/v)甲酸水水溶液为溶剂,配制成1.0、2.0、5.0、10、20、50、100、200ng/ml丙烯酰胺系列标准工作溶液(均含100ng/ml
13
c3d
3-丙烯酰胺)。
[0122]
(2)标准曲线绘制:将丙烯酰胺系列标准工作液按照低浓度到高浓度的顺序分别进样进行lc-ms/ms检测,测得相应的相对峰面积获得线性范围为1~200ng/ml丙烯酰胺(含100ng/ml
13
c3d
3-丙烯酰胺)的标准曲线,以丙烯酰胺标准品的浓度为横坐标(x),以丙烯酰胺标准品与
13
c3d
3-丙烯酰胺的峰面积比为纵坐标(y)绘制标准曲线,标准曲线为:y=0.451215x+0.13012,r=0.9999,丙烯酰胺的标准曲线如图3所示。
[0123]
其中,lc-ms/ms检测中液相色谱检测的条件:色谱柱:chromcore biphenyl色谱柱(3μm,3.0mm
×
150mm);柱温:30℃;进样量为10μl;流动相a为0.1%(v/v)甲酸水溶液,流动相b为甲醇,采用的梯度洗脱程序如表3所示。
[0124]
表3液相色谱梯度洗脱条件
[0125][0126][0127]
lc-ms/ms检测中质谱检测的条件包括:离子源为电喷雾离子源(esi+);离子源温度为150℃;毛细管电压为3.0kv;脱溶剂温度为500℃;扫描方式为正离子扫描;检测方式为多反应检测(mrm);雾化气、锥孔反吹气由氮气发生器产生或使用高纯氮气、碰撞气均为高纯氩气(纯度99.999%),使用前应调节各气体流量以使质谱灵敏度达到检测要求;锥孔电压、碰撞能量等电压值应优化至最优灵敏度;定性离子对、定量离子对,锥孔电压及碰撞能量如表1所示,丙烯酰胺及其内标(
13
c3d
3-丙烯酰胺)离子图谱如图2所示。
[0128]
实施例2
[0129]
快速净化固相萃取柱由柱体、设置在柱体内的两块筛板和装填在两块筛板之间的填料(粒径为10~100μm)组成,填料为质量比为1:1的混合型阴离子交换填料和混合型阳离子交换填料。
[0130]
快速净化固相萃取柱的制备方法包括以下步骤:在柱体内设置下筛板,将填料装填到所述下筛板表面,在所述填料上方设置上筛板,得到快速净化固相萃取柱,其中,填料装填方式1:分层装填,自上而下依次为:混合型阴离子交换填料500mg、混合型阳离子交换填料500mg(记为上阴下阳填料固相萃取柱);填料装填方式2:分层装填,自上而下依次为:混合型阳离子交换填料500mg、混合型阴离子交换填料500mg(记为上阳下阴填料固相萃取柱);填料装填方式3:将500mg混合型阴离子交换填料和500mg混合型阳离子交换填料混合均匀后进行填装(记为阴阳混合填料固相萃取柱)。
[0131]
实施例3
[0132]
(1)样品前处理
[0133]
样品制备:咖啡豆用粉碎机粉碎至粒径为50μm,装于50ml离心管袋中待取样测定。
[0134]
样品提取:准确称取粉碎后的咖啡豆粉或速溶咖啡样品2.0g(精确至0.01g)于15ml刻度离心管中,加入200μl实施例1配制的浓度为5.0mg/l
13
c3d
3-丙烯酰胺内标应用液和10ml水,在室温、2500r/min条件下涡旋提取10min,然后加入5ml二氯甲烷脱脂,在室温、2500r/min条件下涡旋处理5min,在5000r/min条件下离心分离3min,上清液为样品提取液。
[0135]
样品净化:将实施例2制备的固相萃取柱(waters mcx为下层填料,waters max为上层填料)依次用6ml甲醇、10ml水淋洗以活化柱子,取1.0ml所述样品提取液到经活化后的阴阳混合填料固相萃取柱上,待样品全部到填料(筛板表面无样品提取液)中后,用4ml水淋洗除杂,然后用5ml水洗脱并收集,将收集得到的洗脱液转移至50ml离心管中,加入10ml乙腈混匀,加入1g氯化钠和4g无水硫酸镁,室温、2500r/min条件下涡旋提取10min,在5000r/min条件下离心分离5min,取出乙腈层液体,在40℃水浴中氮气吹干,在所得浓缩物加入1.0ml0.1%(v/v)甲酸水溶液进行复溶,过0.22μm滤膜,得到待测样品液。
[0136]
(2)样品的定性和定量检测
[0137]
将待测样品液进样,采用实施例1的lc-ms/ms检测条件进行定性和定量lc-ms/ms检测。
[0138]
(2.1)定性分析:每种待测化合物的质谱定性离子必须出现,至少应包括一个母离子和两个子离子,而且同一检验批次,对同一化合物,样品中目标化合物的两个子离子的相对丰度比与浓度相当的标准溶液相比,其允许偏差不超过表3规定的范围;离子比在
±
20%范围内,可判定为丙烯酰胺阳性。
[0139]
(2.2)定量分析:内标法计算待测液中目标物质的质量浓度,按照前述式(1)计算样品中待测物(丙烯酰胺)的含量,试液中待测物的响应值应在标准曲线线性范围内,超过线性范围则应适当减少取样量后重新测定;
[0140][0141]
式(1)中,x为样品中丙烯酰胺的含量,单位为μg/kg;
[0142]
c为样品中丙烯酰胺按照内标法在标准曲线中对应的浓度,单位为ng/ml;
[0143]
v为样品待测液的定容体积,单位为ml(本发明的实施例中为1ml);
[0144]
f为样品提取液与经快速净化固相萃取柱净化的样品提取液的体积比值;
[0145]
m为样品质量,单位为g。
[0146]
(3)方法精密度实验过程中在样品称量质量后加入相应含量的丙烯酰胺标准使用液,以使得咖啡豆的待测样品液中加标浓度分别为10、20和100μg/kg,速溶咖啡的待测样品液中100、200和500μg/kg,平行测定6次,方法精密度实验结果如表4所示。
[0147]
表4方法精密度实验结果
[0148][0149]
由表4可知,咖啡样品中丙烯酰胺的加标浓度为10~500μg/kg时,平均回收率为87.5~105.3%,精密度(相对标准偏差rsd)为2.6~8.5%。当样品质量为2.0g时,丙烯酰胺的检出限为3μg/kg(以3倍信噪比计算检出限),定量限为10μg/kg(以10倍信噪比计算定量限),说明本发明提供的前处理方法和检测方法的准确度和精确度高、重复性好,适合咖啡豆、咖啡豆粉、速溶咖啡样品的高效快速和准确检测。
[0150]
实施例4
[0151]
市场采集7份咖啡豆、6份咖啡豆粉和10份速溶咖啡,分别按照实施例3中(1)样品前处理的方法进行处理,得到待测样品液,其中,咖啡粉和速溶咖啡不需要进行破碎,测试结果;将待测样品液进样,采用实施例1的lc-ms/ms检测条件进行lc-ms/ms定量检测,实际样品分析结果如表5所示:
[0152]
表5实际样品分析结果
[0153]
[0154][0155]
由表5可知,采集的咖啡豆、咖啡豆粉和速溶咖啡中均含有一定量丙烯酰胺,且部分含量较高,本发明提供的前处理方法和检测方法完全可以满足实际样品检测要求。
[0156]
实施例5
[0157]
按照实施例3步骤(1)~(2)的方法对fapas速溶咖啡质控样品进行测定(编号tyg068rm,该质控样赋值为604
±
45μg/kg,含量范围为559~649μg/kg),独立测定3次,结果为595.7μg/kg、605.1μg/kg和608.9μg/kg,平均结果为603.2μg/kg,平均值为603.2μg/kg,准确度高。
[0158]
图4为fapas速溶咖啡质控样品离子谱图,由图4可知本发明的固相萃取柱和净化方法适用于咖啡样品的快速准确检测。
[0159]
实施例6
[0160]
按照实施例3步骤(1)~(2)的方法对速溶黑咖啡粉样品进行测定,独立测定3次,结果为200.6μg/kg、217.7μg/kg和207.2μg/kg,平均结果为208.5μg/kg,相对标准偏差为4.14%,方法重复性好,。
[0161]
图5为速溶黑咖啡粉样品离子谱图,由图5可知本发明的固相萃取柱和净化方法适用于咖啡样品的快速准确检测。
[0162]
实施例7
[0163]
将黑咖啡和fapas速溶咖啡按照实施例3步骤(1)~(2)的方法进行前处理和检测,平行3次,结果如表6所示。
[0164]
对比例1
[0165]
分别将黑咖啡和fapas速溶咖啡按照gb5009.204-2014《食品安全国家标准食品中丙烯酰胺的测定》第一法采用液相色谱-串联质谱检测咖啡中的丙烯酰胺进行前处理和检测,样品前处理后得到深色的进样溶液而不能得到澄清的进样溶液,平行3次,检测结果如表6所示。
[0166]
表6实施例7与gb5009.204-2014的比较(n=3)
[0167] gb5009.204-2014(μg/kg)实施例7(μg/kg)含量范围(μg/kg)黑咖啡812.2(有干扰,不准确)208.5/fapas速溶咖啡960.7(有干扰,不准确)603.2604
±
45
[0168]
由表6可知,gb5009.204-2014检测方法由于基质干扰太大,目标物干扰大,无法精准测定,不适合咖啡制品中丙烯酰胺的检测。
[0169]
对比例2
[0170]
欧盟bsiso18862:2016采用水为提取溶剂,正己烷脱脂,水相经铁氰化钾试剂沉淀后,再经c18固相萃取柱和离子交换固相萃取柱2次净化,稀释后进液相色谱-串联质谱分析,检测灵敏度相对较低,操作相对繁琐。
[0171]
而采用本发明提供的快速净化固相萃取柱净化后,得到的进样液(即待测样品液)澄清,大幅度降低了目标基质的干扰。
[0172]
对比例3
[0173]
按照实施例6的方法进行检测,与实施例6的区别在于,内标物质为
13c3-丙烯酰胺。
[0174]
采用
13c3-丙烯酰胺作为内标物质时,
[0175]
图6为实施例6中速溶黑咖啡粉样品离子谱图,图7为对比例3的速溶黑咖啡粉样品离子谱图。通过比较图6和图7可知,
13c3-丙烯酰胺通道的峰为干扰物质,当采用
13c3-丙烯酰胺为内标物质时,干扰物和内标保留时间相近,对内标定量准确性造成干扰,从而影响结果的准确性。而本发明以
13
c3d
3-丙烯酰胺作为内标物质,消除了干扰峰对内标物的影响,定量检测准确性高。
[0176]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1