一种测定赛克含量的方法与流程
5μm色谱柱。
11.进一步地,所述供试品为含量为90%-100%的赛克工业品。
12.进一步地,所述供试品溶液的制备方法包括,采用溶剂溶解供试品,得到溶液,将溶液进行过滤,取滤液,即得。
13.进一步地,供试品溶液的制备过程中,所述溶剂选自水、甲醇和乙醇中的至少一种;和/或,所述溶液中供试品的浓度为0.2~0.7mg/ml。
14.进一步地,所述标准品溶液的制备方法包括,采用溶剂溶解赛克标准品,得到溶液,将溶液进行过滤,取滤液,即得。
15.进一步地,标准品溶液制备过程中,所述溶剂选自水、甲醇和乙腈中的至少一种;和/或,所述标准品溶液的浓度为0.02~1mg/ml。
16.进一步地,步骤(1)中配制n个浓度为0.02-1mg/ml的标准品溶液,其中n为≥3的整数;优选地,n为≥5的整数(例如5、6、7、8、9、10),更优选地,n个标准溶液中赛克的浓度分别为:0.02mg/ml、0.1mg/ml、0.2mg/ml、0.5mg/ml和1mg/ml。
17.本发明技术方案,具有如下优点:
18.1.本发明提供的测定赛克含量的方法,通过采用以辛烷基硅烷键合硅胶为填充剂的色谱柱,以乙腈为流动相b,以含磷酸盐的水溶液为流动相a,并采用特定的洗脱程序,不仅实现了赛克工业品中的主成分与其他杂质有效分离,而且峰形对称性良好,不会出现肩峰、分叉峰等现象,可以实现赛克含量的准确测定。
19.2.本发明提供的测定赛克含量的方法,通过控制柱温为25-40℃,尤其是30-40℃,能够进一步改良峰形的对称性和基线的平稳性。
附图说明
20.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
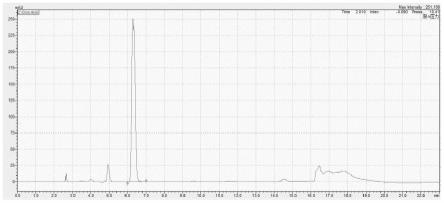
21.图1是实施例1中供试品溶液的色谱图;
22.图2是对比例1中供试品溶液的色谱图;
23.图3是对比例2中供试品溶液的色谱图;
24.图4是对比例3中供试品溶液的色谱图;
25.图5是对比例4中供试品溶液的色谱图。
具体实施方式
26.提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
27.实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得
的常规试剂产品。
28.实施例中使用赛克标准品购自国药集团化学试剂有限公司,纯度为99%;赛克工业品由佳化化学科技发展(上海)有限公司提供。
29.实施例1
30.本实施例提供了一种测定赛克含量的方法,包括如下步骤:
31.(1)分别用超纯水溶解赛克标准品,配制得到浓度分别为0.02、0.1、0.2、0.5、1mg/ml的溶液,过0.45μm滤膜过滤,得到标准品溶液。
32.(2)采用超纯水溶解赛克工业品,得到0.2mg/ml的试样溶液,并将配制好的溶液经过0.45μm滤膜过滤,得到供试品溶液。
33.(3)采用液相色谱法测定标准品溶液和供试品溶液,并根据测试结果制作标准曲线,计算供试品溶液的浓度,色谱条件为:检测波长为:210nm,色谱柱为shim-pack gist c8 5μm;柱温箱40℃;流速1ml/min;流动相:a相:5wt%磷酸二氢钠水溶液,b相:乙腈;梯度洗脱程序如下:0-3min:体积百分数为5%的流动相b,95%流动相a;3-15min:体积百分数为5%的流动相b,体积百分数为95%的流动相a
→
体积百分数为100%的流动相b,体积百分数为0%的流动相a;15-20min:体积百分数为100%的流动相b,体积百分数为0%的流动相a
→
体积百分数为5%的流动相b,体积百分数为95%的流动相a,进样量为50μl。
34.表1标准曲线结果
[0035][0036]
根据表1中结果,以标准品溶液的浓度为横坐标,峰面积为纵坐标进行线性回归,得到回归方程:y=2
×
106x+10944;r2=0.9999。其中y为峰面积,x为浓度。
[0037]
供试品溶液的色谱图见图1所示,赛克出峰时间6.3min,根据供试品的峰面积和回归方程计算供试品中赛克的含量为92%,基线平稳,无肩缝和分裂峰存在,分离度为1.64,拖尾因子为1.02,两者均符合分析要求,测试结果准确可靠。
[0038]
对比例1
[0039]
本实施例提供了一种测定赛克含量的方法,采用液相色谱法检测与实施例1同批次的供试品溶液,色谱条件基本与实施例1相同,区别仅在于柱温不同,本实施例采用柱温为25℃。
[0040]
供试品溶液的色谱图见图2所示,赛克出峰时间6.6min,虽无肩缝和分裂峰存在,但是分离度为1.41,且稍有拖尾,基线稍有不稳,拖尾因子为1.156,不符合分析要求(分离度大于1.5,拖尾因子0.95~1.05之间),根据供试品的峰面积和回归方程计算供试品中赛克的含量为90%。
[0041]
对比例2
[0042]
本对比例提供了一种测定赛克含量的方法,采用液相色谱法检测与实施例1同批次的供试品溶液,色谱条件基本与实施例1相同,区别仅在于流动相不同,本对比例采用超纯水替换实施例1的5%磷酸二氢钠水溶液。
[0043]
供试品溶液的色谱图见图3所示,出现分叉峰,分离度为1.39,不符合分析要求。且
有拖尾,拖尾因子为1.124,不符合液相分析的要求。
[0044]
对比例3
[0045]
本对比例提供了一种测定赛克含量的方法,采用液相色谱法检测与实施例1同批次的供试品溶液,色谱条件基本与实施例1相同,区别仅在于本对比例采用体积比为95:5的含5wt%的磷酸二氢钠水溶液和乙腈为流动相进行等度洗脱。
[0046]
供试品溶液的色谱图见图4所示,赛克峰出现明显拖尾现象,拖尾因子为1.272,不符合液相分析的要求。
[0047]
对比例4
[0048]
采用吴建军在文献《高效液相色谱法检测三羟乙基异氰尿酸酯的纯度》,氨基酸和生物资源,2010(03)中公开的方法测定本发明实施例1中同批次赛克工业品中赛克的含量,结果见图5所示。
[0049]
如图5所示,赛克峰出现明显拖尾现象,拖尾因子为1.294,不符合液相分析的要求。
[0050]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1